Comprehensive Analysis of Hepatitis B Virus Promoter Region Mutations

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
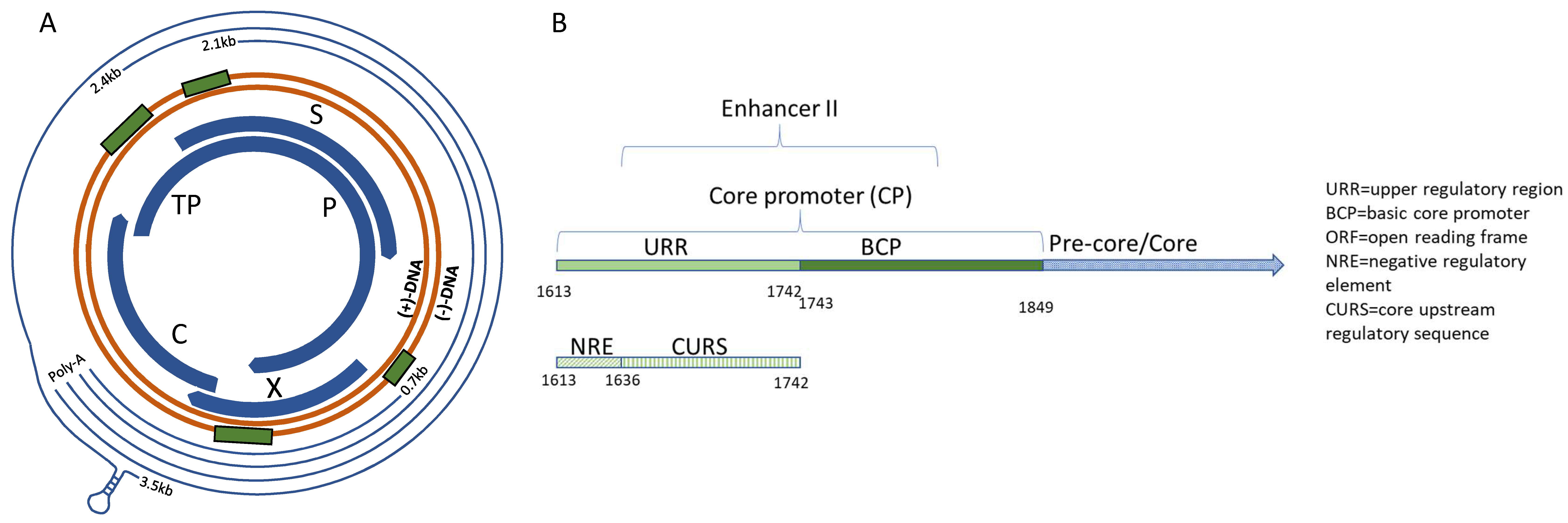
3. Results
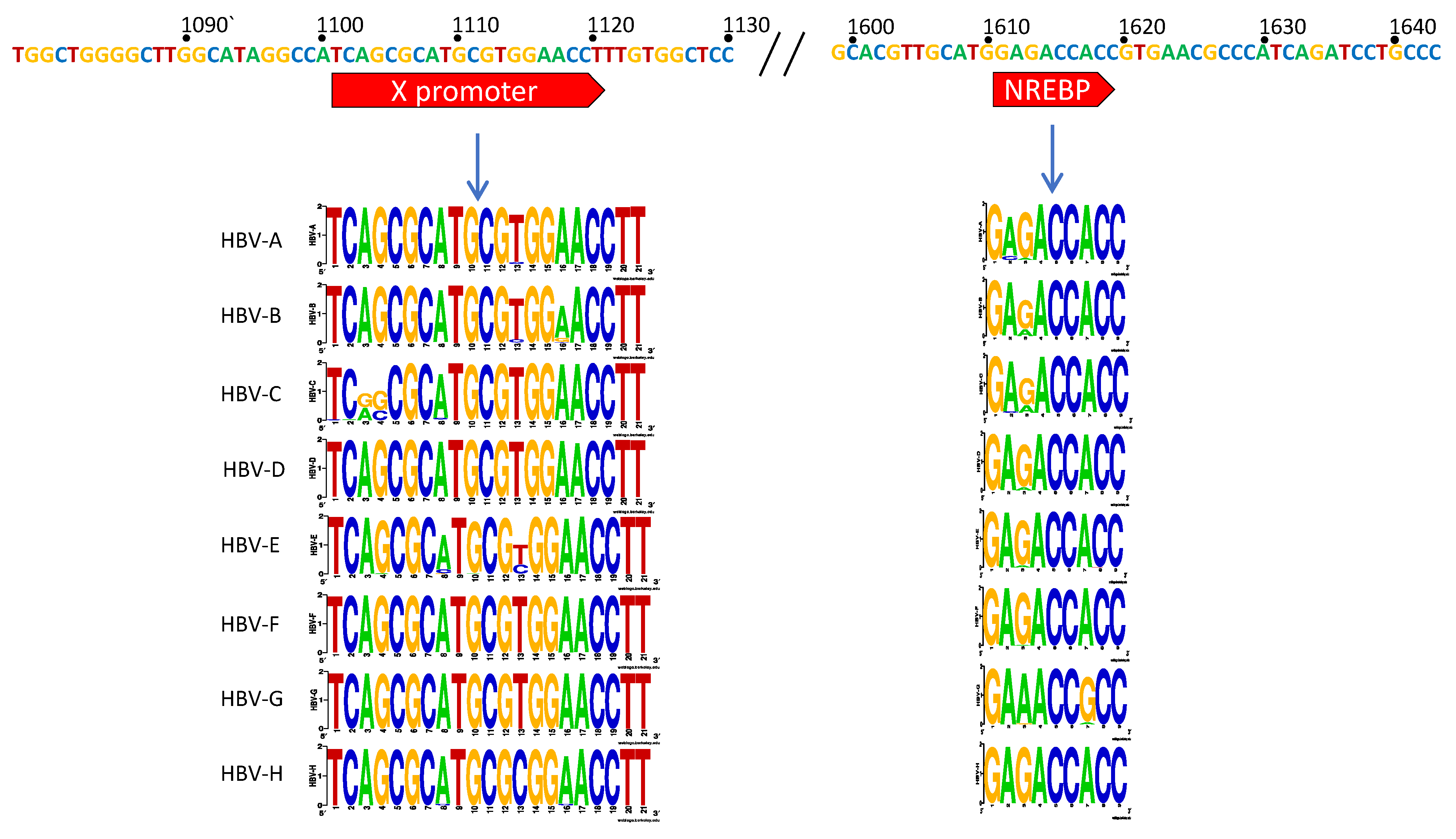
3.1. X Gene Promoter Region
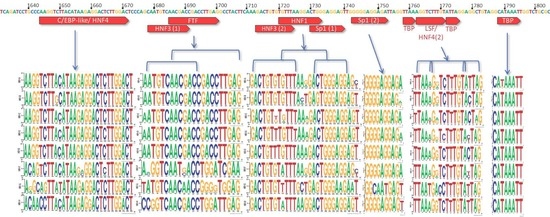
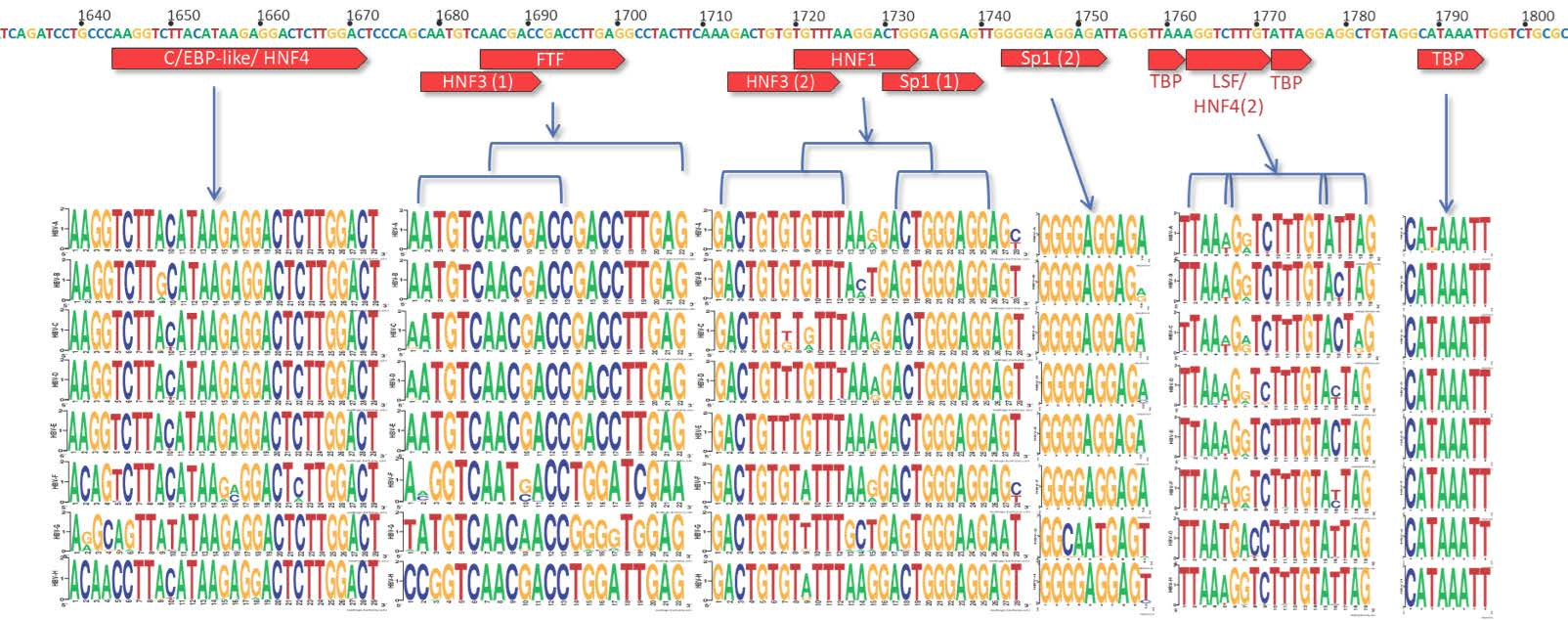
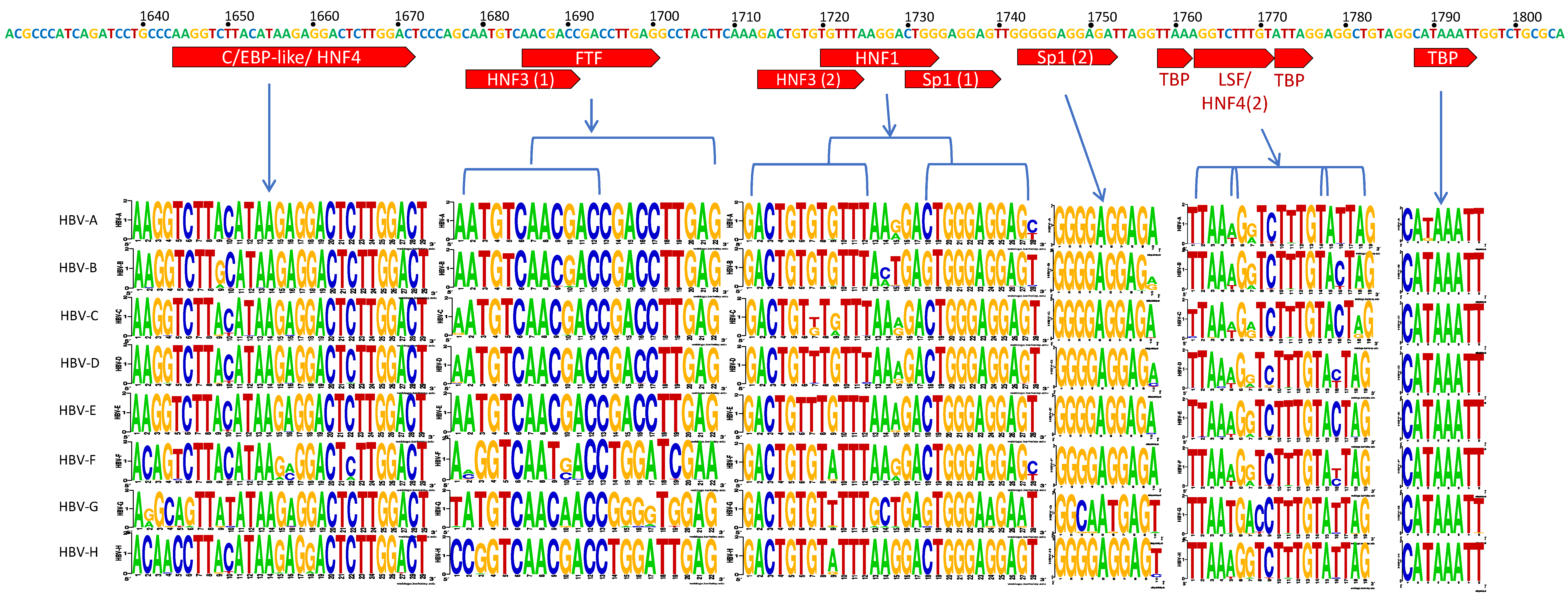
3.2. C Gene/HBV Core Promoter Region
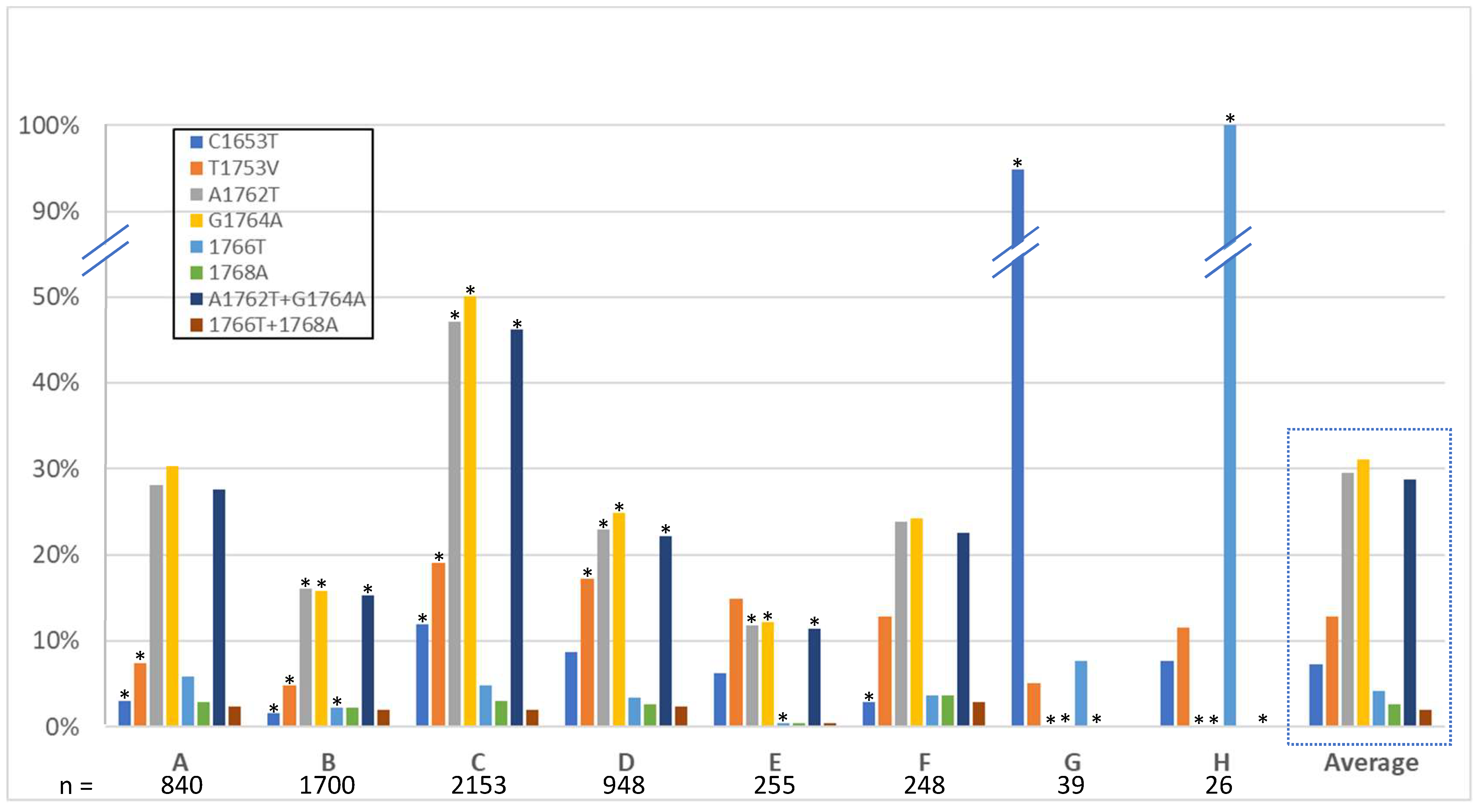
3.3. High-Risk HCC Mutations
4. Discussion
4.1. The X Promoter
4.2. Importance of the C Gene/HBV Core Promoter Region
4.2.1. Genotype G has Many Unique Core Promoter Features
4.2.2. Other Core Promoter Interactions
4.3. High-Risk HCC Mutations
4.4. Study Limitations
4.5. Future Perspectives
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Global Hepatitis Report. 2017. Available online: http://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (accessed on 30 January 2018).
- Yang, H.C.; Kao, J.H. Persistence of hepatitis B virus covalently closed circular DNA in hepatocytes: Molecular mechanisms and clinical significance. Emerg. Microbes Infect. 2014, 3, e64. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamed, A.M.; Kelley, C.M.; Miller, T.G.; Furman, P.A.; Isom, H.C. Rebound of hepatitis B virus replication in HepG2 cells after cessation of antiviral treatment. J. Virol. 2002, 76, 8148–8160. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.M.; Yau, T.O.; Yu, J. Management of chronic hepatitis B infection: Current treatment guidelines, challenges, and new developments. World J. Stroenterol. 2014, 20, 6262–6278. [Google Scholar] [CrossRef] [PubMed]
- Zoulim, F.; Lebosse, F.; Levrero, M. Current treatments for chronic hepatitis B virus infections. Curr. Opin. Virol. 2016, 18, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Arends, J.E.; Lieveld, F.I.; Ahmad, S.; Ustianowski, A. New Viral and Immunological Targets for Hepatitis B Treatment and Cure: A Review. Infect. Dis. Ther. 2017, 6, 461–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durantel, D.; Zoulim, F. New antiviral targets for innovative treatment concepts for hepatitis B virus and hepatitis delta virus. J. Hepatol. 2016, 64, S117–S131. [Google Scholar] [CrossRef] [PubMed]
- Testoni, B.; Durantel, D.; Zoulim, F. Novel targets for hepatitis B virus therapy. Liver Int. 2017, 37, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucifora, J.; Protzer, U. Attacking hepatitis B virus cccDNA—The holy grail to hepatitis B cure. J. Hepatol. 2016, 64, S41–S48. [Google Scholar] [CrossRef] [PubMed]
- Sarah, C. Targeting host kinases. Nat. Rev. Drug Discov. 2017, 16, 314. [Google Scholar] [PubMed]
- Alonso, S.; Guerra, A.R.; Carreira, L.; Ferrer, J.A.; Gutierrez, M.L.; Fernandez-Rodriguez, C.M. Upcoming pharmacological developments in chronic hepatitis B: Can we glimpse a cure on the horizon? BMC Gastroenterol. 2017, 17, 168. [Google Scholar] [CrossRef] [PubMed]
- Périer, R.C.; Junier, T.; Bucher, P. The Eukaryotic Promoter Database EPD. Nucleic Acids Res. 1998, 26, 353–357. [Google Scholar] [CrossRef] [Green Version]
- Dreos, R.; Ambrosini, G.; Cavin Périer, R.; Bucher, P. EPD and EPDnew, high-quality promoter resources in the next-generation sequencing era. Nucleic Acids Res. 2013, 41, D157–D164. [Google Scholar] [CrossRef] [PubMed]
- Promoter Mutations and Cancer. Science 2013, 339, 882. [CrossRef]
- Cuykendall, T.N.; Rubin, M.A.; Khurana, E. Non-coding genetic variation in cancer. Curr. Opin. Syst. Biol. 2017, 1, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.; Guo, X.; Wu, W.; Wang, Y.; Li, G.; Chen, M.; Peng, L.; Wang, S.; Dai, L.; Tang, L.; et al. Integrated analysis of promoter mutation, methylation and expression of AKT1 gene in Chinese breast cancer patients. PLoS ONE 2017, 12, e0174022. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Y.; He, J.; Zhu, M.L.; Teng, X.Y.; Li, Q.X.; Sun, M.H.; Wang, X.F.; Yang, Y.J.; Wang, J.C.; Jin, L.; et al. A Functional Polymorphism (rs2494752) in the AKT1 Promoter Region and Gastric Adenocarcinoma Risk in an Eastern Chinese Population. Sci. Rep. 2016, 6, 20008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griewank, K.G.; Murali, R.; Puig-Butille, J.A.; Schilling, B.; Livingstone, E.; Potrony, M.; Carrera, C.; Schimming, T.; Moller, I.; Schwamborn, M.; et al. TERT promoter mutation status as an independent prognostic factor in cutaneous melanoma. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Xi, L.; Zaug, A.J.; Powell, N.M.; Dancik, G.M.; Cohen, S.B.; Costello, J.C.; Theodorescu, D.; Cech, T.R. Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science 2015, 347, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Kudo, E.; Taura, M.; Suico, M.A.; Goto, H.; Kai, H.; Okada, S. Transcriptional regulation of HIV-1 host factor COMMD1 by the Sp family. Int. J. Mol. Med. 2018, 41, 2366–2374. [Google Scholar] [CrossRef] [PubMed]
- Chong, T.; Chan, W.K.; Bernard, H.U. Transcriptional activation of human papillomavirus 16 by nuclear factor I, AP1, steroid receptors and a possibly novel transcription factor, PVF: A model for the composition of genital papillomavirus enhancers. Nucleic Acids Res. 1990, 18, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Hubert, W.G.; Kanaya, T.; Laimins, L.A. DNA Replication of Human Papillomavirus Type 31 Is Modulated by Elements of the Upstream Regulatory Region That Lie 5′ of the Minimal Origin. J. Virol. 1999, 73, 1835–1845. [Google Scholar] [PubMed]
- Apt, D.; Watts, R.M.; Suske, G.; Bernard, H.U. High Sp1/Sp3 ratios in epithelial cells during epithelial differentiation and cellular transformation correlate with the activation of the HPV-16 promoter. Virology 1996, 224, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 2000, 64, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Pollicino, T.; Petersen, J.; Belloni, L.; Raimondo, G.; Dandri, M. Control of cccDNA function in hepatitis B virus infection. J. Hepatol. 2009, 51, 581–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A.; Bonhoeffer, S.; Hill, A.M.; Boehme, R.; Thomas, H.C.; McDade, H. Viral dynamics in hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 1996, 93, 4398–4402. [Google Scholar] [CrossRef] [PubMed]
- The HBV Database. Available online: https://hbvdb.ibcp.fr/HBVdb/ (accessed on 15 August 2018).
- Hayer, J.; Jadeau, F.; Deléage, G.; Kay, A.; Zoulim, F.; Combet, C. HBVdb: A knowledge database for Hepatitis B Virus. Nucleic Acids Res. 2013, 41, D566–D570. [Google Scholar] [CrossRef] [PubMed]
- Geneious 10.1.3. Available online: www.geneious.com (accessed on 15 August 2018).
- Gunther, S.; Piwon, N.; Iwanska, A.; Schilling, R.; Meisel, H.; Will, H. Type, prevalence, and significance of core promoter/enhancer II mutations in hepatitis B viruses from immunosuppressed patients with severe liver disease. J. Virol. 1996, 70, 8318–8331. [Google Scholar] [PubMed]
- Ishida, H.; Ueda, K.; Ohkawa, K.; Kanazawa, Y.; Hosui, A.; Nakanishi, F.; Mita, E.; Kasahara, A.; Sasaki, Y.; Hori, M.; et al. Identification of Multiple Transcription Factors, HLF, FTF, and E4BP4, Controlling Hepatitis B Virus Enhancer II. J. Virol. 2000, 74, 1241–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raney, A.K.; Johnson, J.L.; Palmer, C.N.; McLachlan, A. Members of the nuclear receptor superfamily regulate transcription from the hepatitis B virus nucleocapsid promoter. J. Virol. 1997, 71, 1058–1071. [Google Scholar] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- WebLogo. Available online: https://weblogo.berkeley.edu/logo.cgi (accessed on 30 August 2018).
- Quarleri, J. Core promoter: A critical region where the hepatitis B virus makes decisions. World J. Gastroenterol. 2014, 20, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Quasdorff, M.; Protzer, U. Control of hepatitis B virus at the level of transcription. J. Viral Hepat. 2010, 17, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Tokusumi, Y.; Zhou, S.; Takada, S. Nuclear respiratory factor 1 plays an essential role in transcriptional initiation from the hepatitis B virus x gene promoter. J. Virol. 2004, 78, 10856–10864. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, Z.; Zheng, Y.; Johnson, D.L.; Ou, J.H. Regulation of hepatocyte nuclear factor 1 activity by wild-type and mutant hepatitis B virus X proteins. J. Virol. 2002, 76, 5875–5881. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Wu, D.; Yang, W.; Weng, M.; Li, Y.; Wang, X.; Zhang, X.; Jin, X.; Wang, T. Hepatitis B virus x protein induces epithelial-mesenchymal transition of hepatocellular carcinoma cells by regulating long non-coding RNA. Virol J. 2017, 14, 238. [Google Scholar] [CrossRef] [PubMed]
- Sekiba, K.; Otsuka, M.; Ohno, M.; Yamagami, M.; Kishikawa, T.; Suzuki, T.; Ishibashi, R.; Seimiya, T.; Tanaka, E.; Koike, K. Hepatitis B virus pathogenesis: Fresh insights into hepatitis B virus RNA. World J. Gastroenterol. 2018, 24, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, J.; Ou, J.H. Regulation of hepatitis B virus core promoter by transcription factors HNF1 and HNF4 and the viral X protein. J. Virol. 2004, 78, 6908–6914. [Google Scholar] [CrossRef] [PubMed]
- Kew, M.C. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2011, 26, 144–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupberger, J.; Hildt, E. Hepatitis B virus-induced oncogenesis. World J. Gastroenterol. 2007, 13, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.T.; Lo, W.Y.; Wang, I.H.; Lo, Y.H.; Shiou, S.R.; Lai, C.K.; Ting, L.P. Transcription repression of human hepatitis B virus genes by negative regulatory element-binding protein/SON. J. Biol. Chem. 2001, 276, 24059–24067. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Chen, M.; Yen, T.S.; Ou, J.H. Hepatocyte-specific expression of the hepatitis B virus core promoter depends on both positive and negative regulation. Mol. Cell. Biol. 1993, 13, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Cabrera, M.; Letovsky, J.; Hu, K.Q.; Siddiqui, A. Transcriptional factor C/EBP binds to and transactivates the enhancer element II of the hepatitis B virus. Virology 1991, 183, 825–829. [Google Scholar] [CrossRef]
- Chen, M.; Hieng, S.; Qian, X.; Costa, R.; Ou, J.H. Regulation of hepatitis B virus ENI enhancer activity by hepatocyte-enriched transcription factor HNF3. Virology 1994, 205, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.-y.; Kim, H.-j.; Park, C.; So, H.-s.; Park, R.K.; Kim, H.C. Impact of Nucleotide Mutations at the HNF3- and HNF4-Binding Sites in Enhancer 1 on Viral Replication in Patients with Chronic Hepatitis B Virus Infection. Gut Liver 2013, 7, 569–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Xie, Y.; Wu, X.; Kong, Y.; Wang, Y. HNF3 binds and activates the second enhancer, ENII, of hepatitis B virus. Virology 1995, 214, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Yuh, C.H.; Chang, Y.L.; Ting, L.P. Transcriptional regulation of precore and pregenomic RNAs of hepatitis B virus. J. Virol. 1992, 66, 4073–4084. [Google Scholar] [PubMed]
- Zhang, P.; McLachlan, A. Differentiation-specific transcriptional regulation of the hepatitis B virus nucleocapsid gene in human hepatoma cell lines. Virology 1994, 202, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Raney, A.K.; McLachlan, A. Characterization of functional Sp1 transcription factor binding sites in the hepatitis B virus nucleocapsid promoter. J. Virol. 1993, 67, 1472–1481. [Google Scholar] [PubMed]
- Liu, S.; Zhang, H.; Gu, C.; Yin, J.; He, Y.; Xie, J.; Cao, G. Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: A meta-analysis. J. Natl. Cancer Inst. 2009, 101, 1066–1082. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.I.; Yeh, S.H.; Chen, P.J.; Iloeje, U.H.; Jen, C.L.; Su, J.; Wang, L.Y.; Lu, S.N.; You, S.L.; Chen, D.S.; et al. Associations between hepatitis B virus genotype and mutants and the risk of hepatocellular carcinoma. J. Natl. Cancer Inst. 2008, 100, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-I.; Yeh, S.-H.; Chen, P.-J.; Iloeje, U.H.; Jen, C.-L.; Su, J.; Wang, L.-Y.; Lu, S.-N.; You, S.-L.; Chen, D.-S.; et al. Associations Between Hepatitis B Virus Genotype and Mutants and the Risk of Hepatocellular Carcinoma. J. Natl. Cancer Inst. 2008, 100, 1134–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, P.; Xu, J.; Yu, Y.; Winkler, C.A. Host and Viral Genetic Variation in HBV-Related Hepatocellular Carcinoma. Front. Genet. 2018, 9, 261. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.L.-Y.; Hui, A.Y.; Wong, M.L.; Tse, A.M.-L.; Hung, L.C.-T.; Wong, V.W.-S.; Sung, J.J.-Y. Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut 2004, 53, 1494–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ching, L.K.; Gounder, P.P.; Bulkow, L.; Spradling, P.R.; Bruce, M.G.; Negus, S.; Snowball, M.; McMahon, B.J. Incidence of hepatocellular carcinoma according to hepatitis B virus genotype in Alaska Native people. Liver Int. 2016, 36, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Bagga, S.; Rawat, S.; Ajenjo, M.; Bouchard, M.J. Hepatitis B virus (HBV) X protein-mediated regulation of hepatocyte metabolic pathways affects viral replication. Virology 2016, 498, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.J.; Schneider, R.J. The Enigmatic X Gene of Hepatitis B Virus. J. Virol. 2004, 78, 12725–12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; You, X.; Li, N.; Zhang, W.; Gagos, S.; Wang, Q.; Banos, A.; Cai, N.; Zhang, H.; Zhang, H.; et al. Involvement of hepatitis B virus X gene (HBx) integration in hepatocarcinogenesis via a recombination of HBx/Alu core sequence/subtelomeric DNA. FEBS Lett. 2012, 586, 3215–3221. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Zheng, Y.; Guo, X.; Wang, Y.; Liu, C. Hepatitis B viral X protein alters the biological features and expressions of DNA repair enzymes in LO2 cells. Liver Int. 2010, 30, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Xiang, T.; Ren, G.; Tan, C.; Liu, R.; Xu, X.; Wu, Z. miR-101 is down-regulated by the hepatitis B virus x protein and induces aberrant DNA methylation by targeting DNA methyltransferase 3A. Cell Signal. 2013, 25, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Yang, W.; Song, J.; Wu, Y.; Ni, B. Hepatitis B virus X protein-induced aberrant epigenetic modifications contributing to human hepatocellular carcinoma pathogenesis. Mol. Cell. Biol. 2013, 33, 2810–2816. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.; Kim, H.S.; Lim, S.O.; Yu, D.Y.; Jung, G. Hepatitis B viral X protein interacts with tumor suppressor adenomatous polyposis coli to activate Wnt/beta-catenin signaling. Cancer Lett. 2011, 300, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Choi, H.S.; Park, Y.K.; Park, E.S.; Shin, G.C.; Kim, D.H.; Ahn, S.H.; Kim, K.H. HBx-induced NF-kappaB signaling in liver cells is potentially mediated by the ternary complex of HBx with p22-FLIP and NEMO. PLoS ONE 2013, 8, e57331. [Google Scholar]
- Luo, J.; Zhou, H.; Wang, F.; Xia, X.; Sun, Q.; Wang, R.; Cheng, B. The hepatitis B virus X protein downregulates NF-kappaB signaling pathways through decreasing the Notch signaling pathway in HBx-transformed L02 cells. Int. J. Oncol. 2013, 42, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Kang, J.A.; Han, M.H.; Chung, K.H.; Lee, C.R.; Song, W.K.; Jun, Y.; Park, S.G. Peroxisome-localized hepatitis Bx protein increases the invasion property of hepatocellular carcinoma cells. Arch. Virol. 2014, 159, 2549–2557. [Google Scholar] [CrossRef] [PubMed]
- Geng, M.; Xin, X.; Bi, L.-Q.; Zhou, L.-T.; Liu, X.-H. Molecular mechanism of hepatitis B virus X protein function in hepatocarcinogenesis. World J. Gastroenterol. 2015, 21, 10732–10738. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef] [PubMed]
- Buti, M.; Rodriguez-Frias, F.; Jardi, R.; Esteban, R. Hepatitis B virus genome variability and disease progression: The impact of pre-core mutants and HBV genotypes. J. Clin. Virol. 2005, 34, 79S–S82. [Google Scholar] [CrossRef]
- Parekh, S.; Zoulim, F.; Ahn, S.H.; Tsai, A.; Li, J.; Kawai, S.; Khan, N.; Trepo, C.; Wands, J.; Tong, S. Genome replication, virion secretion, and e antigen expression of naturally occurring hepatitis B virus core promoter mutants. J. Virol. 2003, 77, 6601–6612. [Google Scholar] [CrossRef] [PubMed]
- Jammeh, S.; Tavner, F.; Watson, R.; Thomas, H.C.; Karayiannis, P. Effect of basal core promoter and pre-core mutations on hepatitis B virus replication. J. Gen. Virol. 2008, 89, 901–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriyama, K.; Okamoto, H.; Tsuda, F.; Mayumi, M. Reduced precore transcription and enhanced core-pregenome transcription of hepatitis B virus DNA after replacement of the precore-core promoter with sequences associated with e antigen-seronegative persistent infections. Virology 1996, 226, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Buckwold, V.E.; Xu, Z.; Chen, M.; Yen, T.S.; Ou, J.H. Effects of a naturally occurring mutation in the hepatitis B virus basal core promoter on precore gene expression and viral replication. J. Virol. 1996, 70, 5845–5851. [Google Scholar] [PubMed]
- Li, K.; Zoulim, F.; Pichoud, C.; Kwei, K.; Villet, S.; Wands, J.; Li, J.; Tong, S. Critical role of the 36-nucleotide insertion in hepatitis B virus genotype G in core protein expression, genome replication, and virion secretion. J. Virol. 2007, 81, 9202–9215. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, M.; Zorgdrager, F.; Bruisten, S.M.; Bakker, M.; Berkhout, B.; van der Kuyl, A.C. Widespread hepatitis B virus genotype G (HBV-G) infection during the early years of the HIV epidemic in the Netherlands among men who have sex with men. BMC Infect. Dis. 2016, 16, 268. [Google Scholar] [CrossRef] [PubMed]
- Van der Kuyl, A.C.; Zorgdrager, F.; Hogema, B.; Bakker, M.; Jurriaans, S.; Back, N.K.; Berkhout, B.; Zaaijer, H.L.; Cornelissen, M. High prevalence of hepatitis B virus dual infection with genotypes A and G in HIV-1 infected men in Amsterdam, The Netherlands, during 2000–2011. BMC Infect. Dis. 2013, 13, 540. [Google Scholar] [CrossRef] [PubMed]
- Osiowy, C.; Gordon, D.; Borlang, J.; Giles, E.; Villeneuve, J.P. Hepatitis B virus genotype G epidemiology and co-infection with genotype A in Canada. J. Gen. Virol. 2008, 89, 3009–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chudy, M.; Schmidt, M.; Czudai, V.; Scheiblauer, H.; Nick, S.; Mosebach, M.; Hourfar, M.K.; Seifried, E.; Roth, W.K.; Grunelt, E.; et al. Hepatitis B virus genotype G monoinfection and its transmission by blood components. Hepatology 2006, 44, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaaijer, H.L.; Boot, H.J.; van Swieten, P.; Koppelman, M.H.; Cuypers, H.T. HBsAg-negative mono-infection with hepatitis B virus genotype G. J. Viral Hepat. 2011, 18, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Tsuda, F.; Akahane, Y.; Sugai, Y.; Yoshiba, M.; Moriyama, K.; Tanaka, T.; Miyakawa, Y.; Mayumi, M. Hepatitis B virus with mutations in the core promoter for an e antigen-negative phenotype in carriers with antibody to e antigen. J. Virol. 1994, 68, 8102–8110. [Google Scholar] [PubMed]
- Kato, H.; Orito, E.; Gish, R.G.; Bzowej, N.; Newsom, M.; Sugauchi, F.; Suzuki, S.; Ueda, R.; Miyakawa, Y.; Mizokami, M. Hepatitis B e antigen in sera from individuals infected with hepatitis B virus of genotype G. Hepatology 2002, 35, 922–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehermann, B.; Bertoletti, A. Immunological aspects of antiviral therapy of chronic hepatitis B virus and hepatitis C virus infections. Hepatology 2015, 61, 712–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, B.; Zhang, B.; Wang, L.; Ma, C.; Liu, X.; Zhao, Y.; Jiao, Y. Hepatitis B Virus e Antigen Regulates Monocyte Function and Promotes B Lymphocyte Activation. Viral Immunol. 2017, 30, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Wan, P.; Cao, Y.; Zhang, W.; Chen, J.; Tan, L.; Wang, Y.; Sun, Z.; Zhang, Q.; Wan, Y.; et al. Hepatitis B Virus e Antigen Activates the Suppressor of Cytokine Signaling 2 to Repress Interferon Action. Sci. Rep. 2017, 7, 1729. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Orito, E.; Gish, R.G.; Sugauchi, F.; Suzuki, S.; Ueda, R.; Miyakawa, Y.; Mizokami, M. Characteristics of Hepatitis B Virus Isolates of Genotype G and Their Phylogenetic Differences from the Other Six Genotypes (A through F). J. Virol. 2002, 76, 6131–6137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, M.; Tanaka, Y.; Sakamoto, T.; Maruyama, I.; Shimada, T.; Takahashi, S.; Shirai, T.; Kato, H.; Nagao, M.; Miyakawa, Y.; et al. Early dynamics of hepatitis B virus in chimeric mice carrying human hepatocytes monoinfected or coinfected with genotype G. Hepatology 2007, 45, 929–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, A.; Kremsdorf, D.; Capel, F.; Housset, C.; Dauguet, C.; Petit, M.A.; Brechot, C. Emergence of and takeover by hepatitis B virus (HBV) with rearrangements in the pre-S/S and pre-C/C genes during chronic HBV infection. J. Virol. 1991, 65, 3566–3574. [Google Scholar] [PubMed]
- Lin, W.J.; Li, J.; Lee, Y.F.; Yeh, S.D.; Altuwaijri, S.; Ou, J.H.; Chang, C. Suppression of hepatitis B virus core promoter by the nuclear orphan receptor TR4. J. Biol. Chem. 2003, 278, 9353–9360. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Mertz, J.E. Differential regulation of the pre-C and pregenomic promoters of human hepatitis B virus by members of the nuclear receptor superfamily. J. Virol. 1997, 71, 9366–9374. [Google Scholar] [PubMed]
- Papatheodoridis, G.V.; Lampertico, P.; Manolakopoulos, S.; Lok, A. Incidence of hepatocellular carcinoma in chronic hepatitis B patients receiving nucleos(t)ide therapy: A systematic review. J. Hepatol. 2010, 53, 348–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meier-Stephenson, V.; Bremner, W.T.R.; Dalton, C.S.; Van Marle, G.; Coffin, C.S.; Patel, T.R. Comprehensive Analysis of Hepatitis B Virus Promoter Region Mutations. Viruses 2018, 10, 603. https://doi.org/10.3390/v10110603
Meier-Stephenson V, Bremner WTR, Dalton CS, Van Marle G, Coffin CS, Patel TR. Comprehensive Analysis of Hepatitis B Virus Promoter Region Mutations. Viruses. 2018; 10(11):603. https://doi.org/10.3390/v10110603
Chicago/Turabian StyleMeier-Stephenson, Vanessa, William T. R. Bremner, Chimone S. Dalton, Guido Van Marle, Carla S. Coffin, and Trushar R. Patel. 2018. "Comprehensive Analysis of Hepatitis B Virus Promoter Region Mutations" Viruses 10, no. 11: 603. https://doi.org/10.3390/v10110603
APA StyleMeier-Stephenson, V., Bremner, W. T. R., Dalton, C. S., Van Marle, G., Coffin, C. S., & Patel, T. R. (2018). Comprehensive Analysis of Hepatitis B Virus Promoter Region Mutations. Viruses, 10(11), 603. https://doi.org/10.3390/v10110603