TIM-1 Promotes Japanese Encephalitis Virus Entry and Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus Preparation
2.2. Virus Challenge and Titration
2.3. Plasmid Construction and Antibodies
2.4. RNA Interference and Plasmid Transfection
2.5. Virus Attachment and Entry Assays
2.6. Real-Time Polymerase Chain Reaction (PCR)
2.7. Western Blot Analysis
2.8. Confocal Microscopy
2.9. Statistical Analysis
3. Results
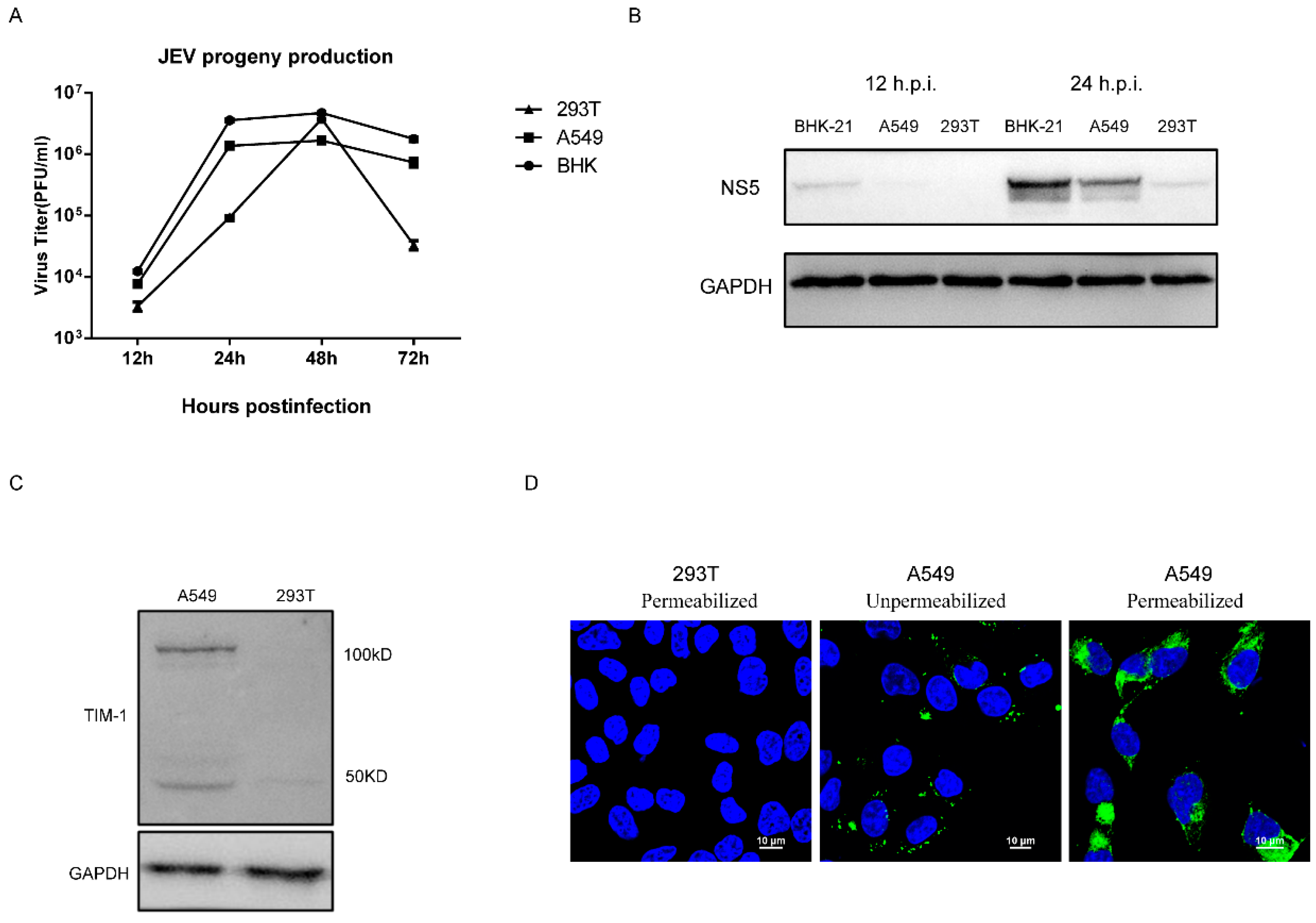
3.1. 293T Cells Are Poorly Permissive to JEV Infection at Early Stages
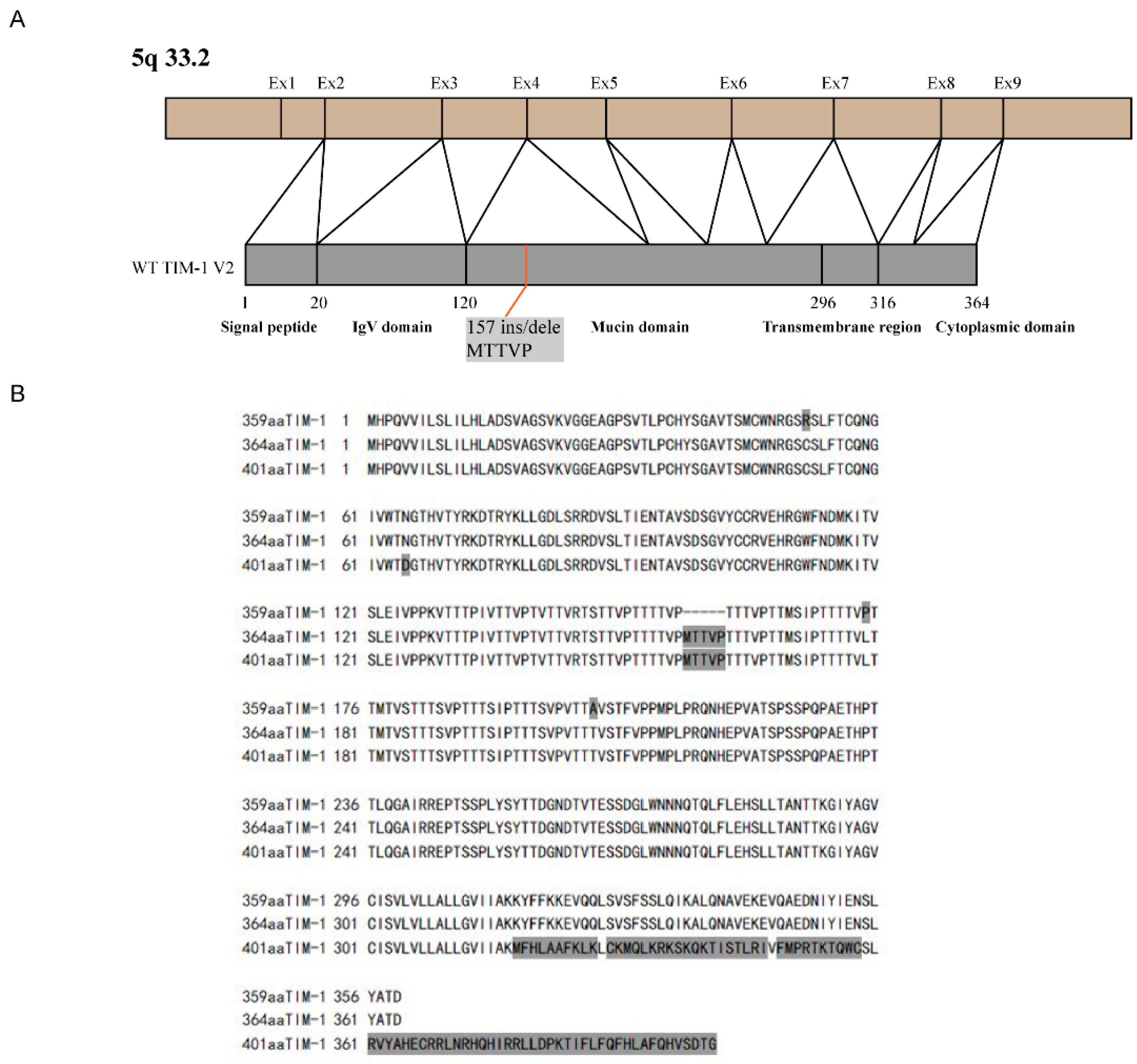
3.2. Human TIM-1 Gene Clone and Polymorphism Analysis
3.3. Ectopic TIM-1 V2 (364aa) Expression in 293T Cells Markedly Enhances JEV Infection
3.4. TIM-1 Promotes JEV Attachment and Internalization, and May Be Associated with the Intracellular Trafficking of Virions
3.5. Phosphatidylserine-Binding Capability of TIM-1 is Crucial for Mediating JEV Attachment, Entry, and Infection
3.6. The Cytoplasmic Domain of TIM-1 Is Important for Enhancing JEV Entry
3.7. Knock-Down of TIM-1 in A549 Cells Impaired JEV Entry and Infection
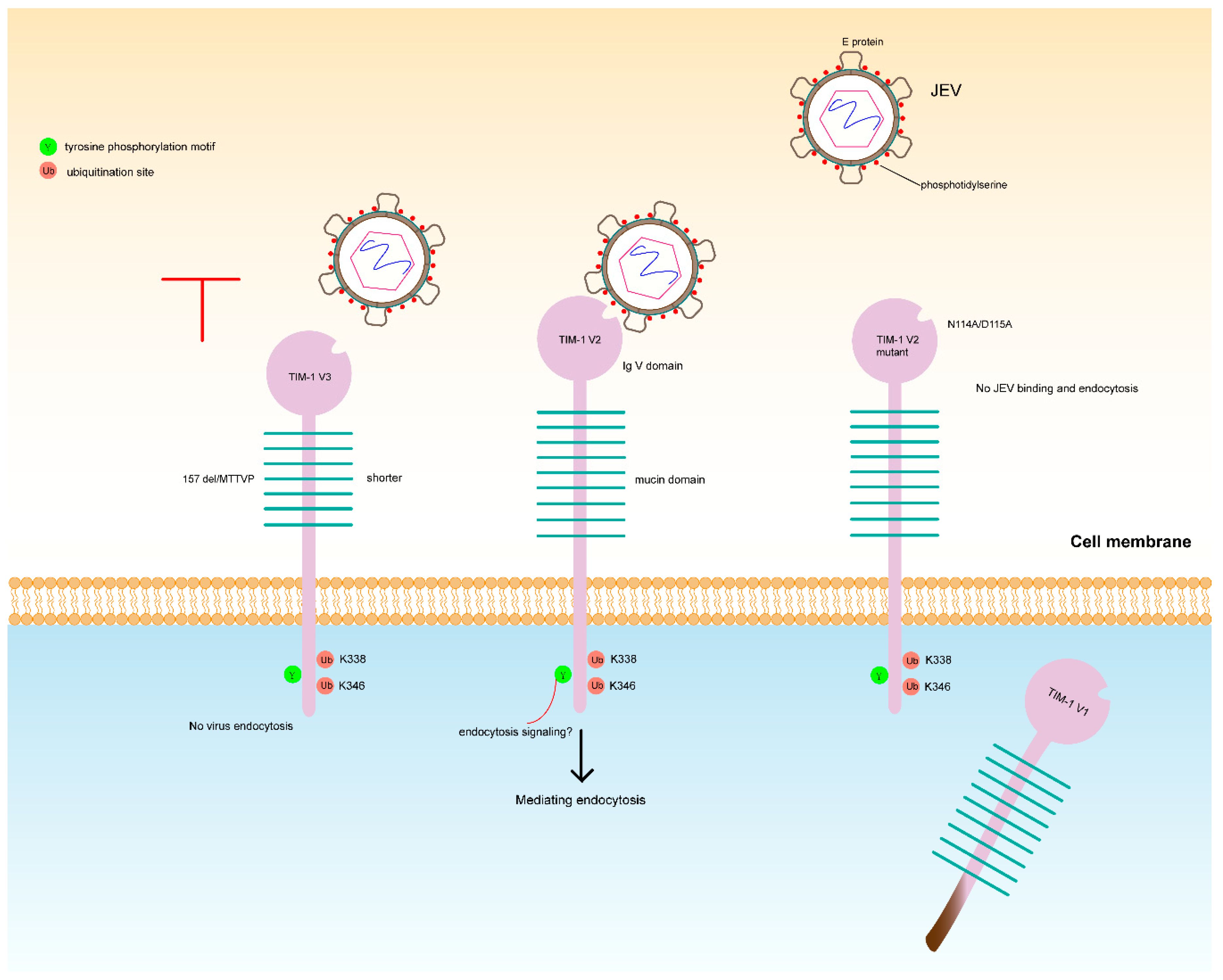
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Williams, M.; Lopez, A.L.; Aldaba, J.G.; Roque, V.G.; Tandoc, A.O.; Sy, A.K.; Espino, F.E.; DeQuiroz-Castro, M.; Jee, Y.; Ducusin, M.J.; et al. Epidemiology of Japanese encephalitis in the Philippines: A systematic review. PLoS Negl. Trop. Dis. 2015, 9, e0003630. [Google Scholar] [CrossRef]
- Wang, H.Y.; Takasaki, T.; Fu, S.H.; Sun, X.H.; Zhang, H.L.; Wang, Z.X.; Hao, Z.Y.; Zhang, J.K.; Tang, Q.; Kotaki, A.; et al. Molecular epidemiological analysis of Japanese encephalitis virus in China. J. Gen. Virol. 2007, 88, 885–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Hurk, A.F.; Ritchie, S.A.; Mackenzie, J.S. Ecology and geographical expansion of Japanese encephalitis virus. Annu. Rev. Entomol. 2009, 54, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Luca, V.C.; AbiMansour, J.; Nelson, C.A.; Fremont, D.H. Crystal structure of the Japanese encephalitis virus envelope protein. J. Virol. 2011, 86, 2337–2346. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Kielian, M. Flaviviruses: Braking the entering. Curr. Opin. Virol. 2013, 3, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Perera-Lecoin, M.; Meertens, L.; Carnec, X.; Amara, A. Flavivirus entry receptors: An update. Viruses 2013, 6, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Cun, W.; Wu, X.; Shi, Q.; Tang, H.; Luo, G. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J. Virol. 2012, 86, 7256–7267. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Pavy, M.; Young, N.; Freeman, C.; Lobigs, M. Antiviral effect of the heparan sulfate mimetic, PI-88, against dengue and encephalitic flaviviruses. Antivir. Res. 2006, 69, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.-J.; Chen, W.-J.; Hsu, W.-L.; Chiou, S.-S. Bovine lactoferrin inhibits Japanese encephalitis virus by binding to heparan sulfate and receptor for low density lipoprotein. Virology 2008, 379, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Hu, K.; Luo, S.; Zhang, M.; Deng, X.; Li, C.; Jin, W.; Hu, B.; He, S.; Li, M.; et al. DC-SIGN as an attachment factor mediates Japanese encephalitis virus infection of human dendritic cells via interaction with a single high-mannose residue of viral E glycoprotein. Virology 2016, 488, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Qian, P.; Wang, D.; Zhi, X.; Wei, Y.; Chen, H.; Li, X. Integrin alphavbeta3 promotes infection by Japanese encephalitis virus. Res. Vet. Sci. 2017, 111, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Medigeshi, G.R.; Hirsch, A.J.; Streblow, D.N.; Nikolich-Zugich, J.; Nelson, J.A. West nile virus entry requires cholesterol-rich membrane microdomains and is independent of alpha v beta 3 integrin. J. Virol. 2008, 82, 5212–5219. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Laxminarayana, S.V.; Chandra, N.; Ravi, V.; Desai, A. Heat shock protein 70 on Neuro2a cells is a putative receptor for Japanese encephalitis virus. Virology 2009, 385, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Ding, T. Heat shock protein 90beta in the Vero cell membrane binds Japanese encephalitis virus. Int. J. Mol. Med. 2017, 40, 474–482. [Google Scholar] [CrossRef] [PubMed]
- McIntire, J.J.; Umetsu, D.T.; DeKruyff, R.H. TIM-1, a novel allergy and asthma susceptibility gene. Springer Semin. Immunopathol. 2004, 25, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Jemielity, S.; Wang, J.J.; Chan, Y.K.; Ahmed, A.A.; Li, W.; Monahan, S.; Bu, X.; Farzan, M.; Freeman, G.J.; Umetsu, D.T.; et al. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog. 2013, 9, e1003232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller-Tank, S.; Kondratowicz, A.S.; Davey, R.A.; Rennert, P.D.; Maury, W. Role of the phosphatidylserine receptor TIM-1 in enveloped-virus entry. J. Virol. 2013, 87, 8327–8341. [Google Scholar] [CrossRef] [PubMed]
- Kondratowicz, A.S.; Lennemann, N.J.; Sinn, P.L.; Davey, R.A.; Hunt, C.L.; Moller-Tank, S.; Meyerholz, D.K.; Rennert, P.; Mullins, R.F.; Brindley, M.; et al. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci. USA 2011, 108, 8426–8431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meertens, L.; Carnec, X.; Lecoin, M.P.; Ramdasi, R.; Guivel-Benhassine, F.; Lew, E.; Lemke, G.; Schwartz, O.; Amara, A. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 2012, 12, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika virus infection in human skin cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Belen Eyheramonho, M.; Pichavant, M.; Gonzalez Cambaceres, C.; Matangkasombut, P.; Cervio, G.; Kuperman, S.; Moreiro, R.; Konduru, K.; Manangeeswaran, M.; et al. A polymorphism in TIM1 is associated with susceptibility to severe hepatitis A virus infection in humans. J. Clin. Investig. 2011, 121, 1111–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Qiao, L.; Hou, Z.; Luo, G. TIM-1 promotes hepatitis C virus cell attachment and infection. J. Virol. 2017, 91, e01583-16. [Google Scholar] [CrossRef] [PubMed]
- Umetsu, D.T.; Umetsu, S.E.; Freeman, G.J.; DeKruyff, R.H. TIM gene family and their role in atopic diseases. In Immunology, Phenotype First: How Mutations Have Established New Principles and Pathways in Immunology; Beutler, B., Ed.; Springer: Berlin, Germany, 2008; Volume 321, pp. 201–215. [Google Scholar]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Beginnings of a good apoptotic meal: The find-me and eat-me signaling pathways. Immunity 2011, 35, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Laliberte, J.P.; Moss, B. Appraising the apoptotic mimicry model and the role of phospholipids for poxvirus entry. Proc. Natl. Acad. Sci. USA 2009, 106, 17517–17521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, J.; Helenius, A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 2008, 320, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Casasnovas, J.M.; Umetsu, D.T.; DeKruyff, R.H. TIM genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 2010, 235, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Cao, L.; Ling, H.; Dang, M.; Sun, Y.; Zhang, X.; Chen, Y.; Zhang, L.; Su, D.; Wang, X.; et al. TIM-1 acts a dual-attachment receptor for Ebolavirus by interacting directly with viral GP and the PS on the viral envelope. Protein Cell 2015, 6, 814–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailly, V.; Zhang, Z.; Meier, W.; Cate, R.; Sanicola, M.; Bonventre, J.V. Shedding of kidney injury molecule-1, a putative adhesion protein involved in renal regeneration. J. Biol. Chem. 2002, 277, 39739–39748. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, M.; Agnihotry, S.; Srivastava, A.; Bolia, R.; Yachha, S.K.; Aggarwal, R. Relationship of severity of hepatitis a with polymorphisms in hepatitis a virus cellular receptor 1 (HAVCR1) gene. Ann. Hepatol. 2018, 17, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Biasin, M.; Sironi, M.; Saulle, I.; Pontremoli, C.; Garziano, M.; Cagliani, R.; Trabattoni, D.; Lo Caputo, S.; Vichi, F.; Mazzotta, F.; et al. A 6-amino acid insertion/deletion polymorphism in the mucin domain of TIM-1 confers protections against HIV-1 infection. Microbes Infect. 2017, 19, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Fujikura, D.; Noyori, O.; Kajihara, M.; Maruyama, J.; Miyamoto, H.; Yoshida, R.; Takada, A. A polymorphism of the TIM-1 IgV domain: Implications for the susceptibility to filovirus infection. Biochem. Biophys. Res. Commun. 2014, 455, 223–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.W.; Nguyen, H.Y.; Hanna, S.L.; Sanchez, M.D.; Doms, R.W.; Pierson, T.C. West Nile virus discriminates between DC-SIGN and DC-SIGNR for cellular attachment and infection. J. Virol. 2006, 80, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Dejarnac, O.; Hafirassou, M.L.; Chazal, M.; Versapuech, M.; Gaillard, J.; Perera-Lecoin, M.; Umana-Diaz, C.; Bonnet-Madin, L.; Carnec, X.; Tinevez, J.Y.; et al. TIM-1 ubiquitination mediates dengue virus entry. Cell Rep. 2018, 23, 1779–1793. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ablan, S.D.; Miao, C.; Zheng, Y.M.; Fuller, M.S.; Rennert, P.D.; Maury, W.; Johnson, M.C.; Freed, E.O.; Liu, S.L. TIM-family proteins inhibit HIV-1 release. Proc. Natl. Acad. Sci. USA 2014, 111, E3699–E3707. [Google Scholar] [CrossRef] [PubMed]
- Feigelstock, D.; Thompson, P.; Mattoo, P.; Zhang, Y.; Kaplan, G.G. The human homolog of HAVcr-1 codes for a hepatitis A virus cellular receptor. J. Virol. 1998, 72, 6621–6628. [Google Scholar] [PubMed]
- Kaplan, G.; Totsuka, A.; Thompson, P.; Akatsuka, T.; Moritsugu, Y.; Feinstone, S.M. Identification of a surface glycoprotein on African green monkey kidney cells as a receptor for hepatitis A virus. EMBO J. 1996, 15, 4282–4296. [Google Scholar] [CrossRef] [PubMed]
- Moller-Tank, S.; Maury, W. Phosphatidylserine receptors: Enhancers of enveloped virus entry and infection. Virology 2014, 468–470, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, M.D.; Meertens, L.; Bonazzi, M.; Cossart, P.; Arenzana-Seisdedos, F.; Amara, A. Appraising the roles of CBLL1 and the ubiquitin/proteasome system for flavivirus entry and replication. J. Virol. 2011, 85, 2980–2989. [Google Scholar] [CrossRef] [PubMed]
- Morizono, K.; Chen, I.S. Role of phosphatidylserine receptors in enveloped virus infection. J. Virol. 2014, 88, 4275–4290. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Kota, S.K.; Kuchroo, V.K.; Humphreys, B.D.; Strom, T.B. TIM family proteins promote the lysosomal degradation of the nuclear receptor NUR77. Sci. Signal. 2012, 5, ra90. [Google Scholar] [CrossRef] [PubMed]
- Hunt, C.L.; Lennemann, N.J.; Maury, W. Filovirus entry: A novelty in the viral fusion world. Viruses 2012, 4, 258–275. [Google Scholar] [CrossRef] [PubMed]
- Bressanelli, S.; Stiasny, K.; Allison, S.L.; Stura, E.A.; Duquerroy, S.; Lescar, J.; Heinz, F.X.; Rey, F.A. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 2004, 23, 728–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. Structure of the dengue virus envelope protein after membrane fusion. Nature 2004, 427, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Fujikura, D.; Nanbo, A.; Marzi, A.; Noyori, O.; Kajihara, M.; Maruyama, J.; Matsuno, K.; Miyamoto, H.; Yoshida, R.; et al. Interaction between TIM-1 and NPC1 is important for cellular entry of Ebola virus. J. Virol. 2015, 89, 6481–6493. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Maguire, T.; Marks, R.M. Demonstration of binding of dengue virus envelope protein to target cells. J. Virol. 1996, 70, 8765–8772. [Google Scholar] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, J.; Jiang, Y.; Xu, H.; Zhao, C.; Zhou, G.; Chen, P.; Cao, R. TIM-1 Promotes Japanese Encephalitis Virus Entry and Infection. Viruses 2018, 10, 630. https://doi.org/10.3390/v10110630
Niu J, Jiang Y, Xu H, Zhao C, Zhou G, Chen P, Cao R. TIM-1 Promotes Japanese Encephalitis Virus Entry and Infection. Viruses. 2018; 10(11):630. https://doi.org/10.3390/v10110630
Chicago/Turabian StyleNiu, Jichen, Ya Jiang, Hao Xu, Changjing Zhao, Guodong Zhou, Puyan Chen, and Ruibing Cao. 2018. "TIM-1 Promotes Japanese Encephalitis Virus Entry and Infection" Viruses 10, no. 11: 630. https://doi.org/10.3390/v10110630
APA StyleNiu, J., Jiang, Y., Xu, H., Zhao, C., Zhou, G., Chen, P., & Cao, R. (2018). TIM-1 Promotes Japanese Encephalitis Virus Entry and Infection. Viruses, 10(11), 630. https://doi.org/10.3390/v10110630