Metatranscriptomic Analysis and In Silico Approach Identified Mycoviruses in the Arbuscular Mycorrhizal Fungus Rhizophagus spp.

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant and Fungal Material
2.2. High-Throughput Sequencing
2.3. Sequence Analysis
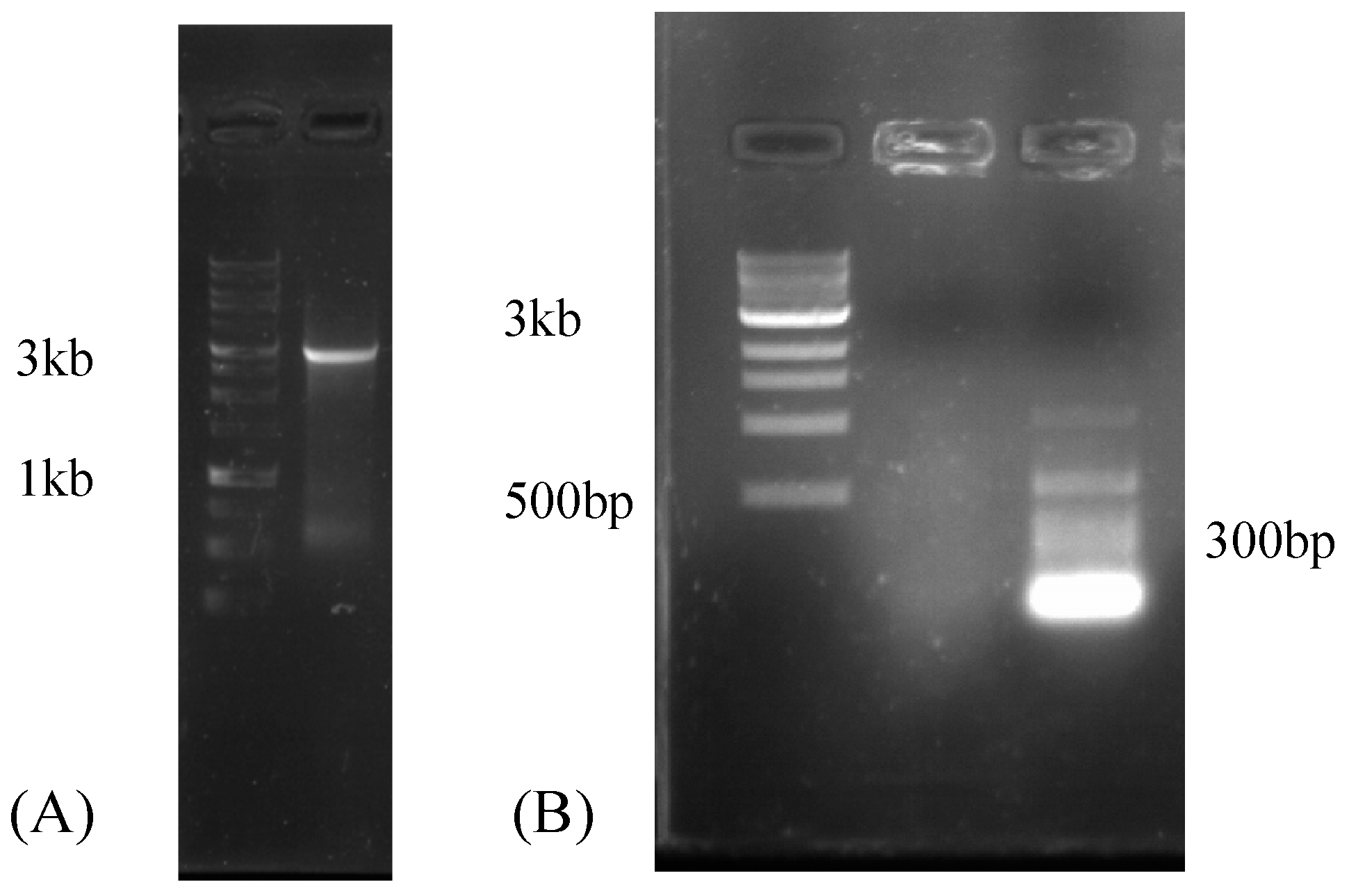
2.4. Reverse-Transcription PCR (RT-PCR)
2.5. Rapid Amplification of cDNA Ends (RACE)
3. Results
3.1. Mycoviruses in the Metatranscriptome of Rhizophagus Irregularis Inoculated Roots
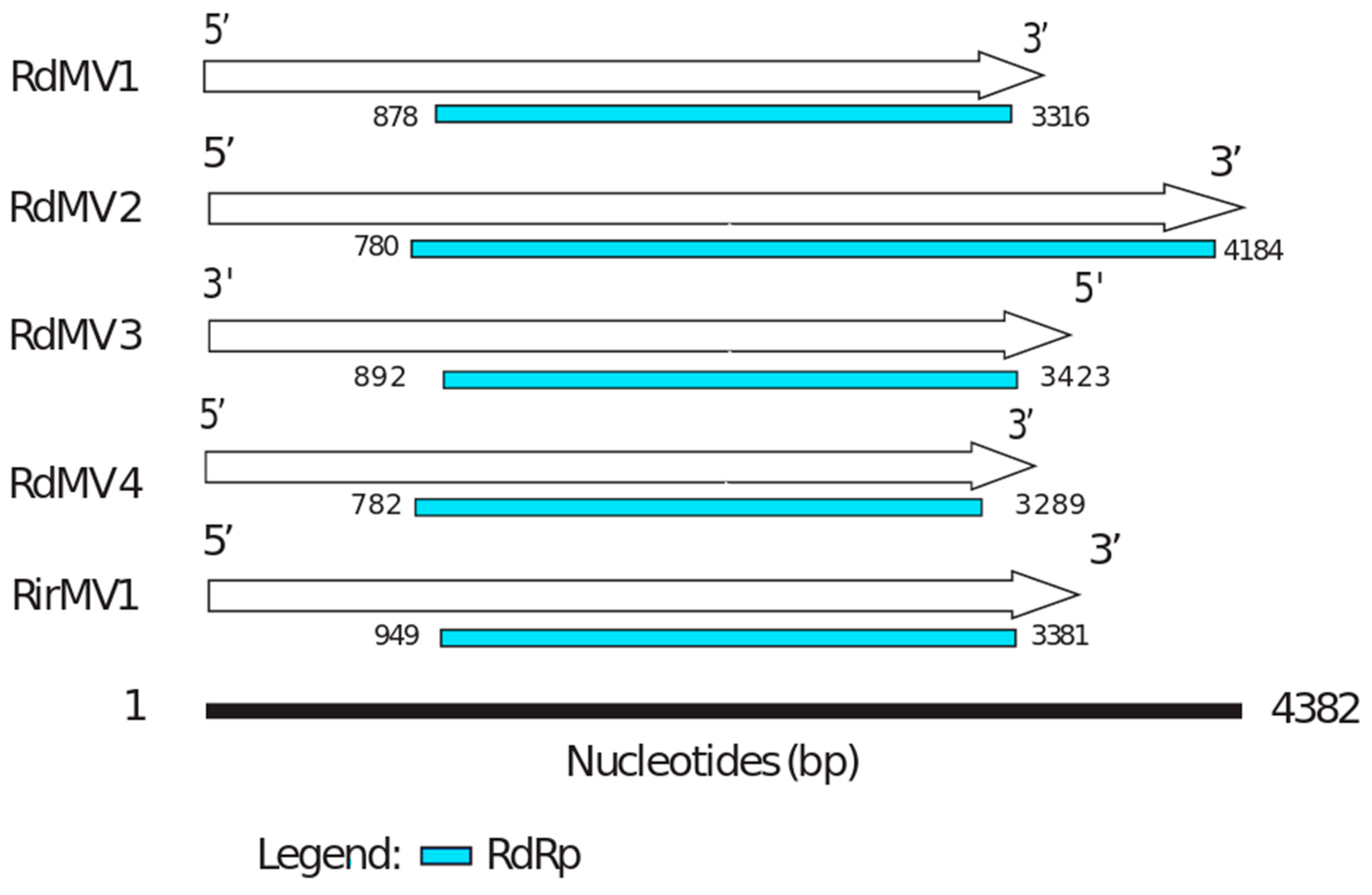
3.2. Mycoviruses in the Transcriptomes of Rhizophagus spp.
3.3. Phylogenetic Analysis and the Characterization of Conserved RdRp Region of Mitoviruses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Smith, S.; Read, D. Mycorrhizal Symbiosis; Academic: San Diego, CA, USA, 1997. [Google Scholar]
- Azcón-Aguilar, C.; Barea, J. Arbuscular mycorrhizas and biological control of soil-borne plant pathogens—An overview of the mechanisms involved. Mycorrhiza 1997, 6, 457–464. [Google Scholar] [CrossRef]
- Augé, R.M. Water relations, drought and vesicular-arbuscular mycorrhizal symbiosis. Mycorrhiza 2001, 11, 3–42. [Google Scholar] [CrossRef]
- Porcel, R.; Aroca, R.; Ruiz-Lozano, J.M. Salinity stress alleviation using arbuscular mycorrhizal fungi. A review. Agron. Sustain. Dev. 2012, 32, 181–200. [Google Scholar] [CrossRef]
- SCHÜßLER, A.; Daniel, S.; Christopher, W. A new fungal phylum, the Glomeromycota: Phylogeny and evolution. Mycol. Res. 2001, 105, 1413–1421. [Google Scholar] [CrossRef]
- Hildebrandt, U.; Kaldorf, M.; Bothe, H. The Zinc Violet and its Colonization by Arbuscular Mycorrhizal Fungi. J. Plant Physiol. 1999, 154, 709–717. [Google Scholar] [CrossRef]
- Kitahara, R.; Ikeda, Y.; Shimura, H.; Masuta, C.; Ezawa, T. A unique mitovirus from Glomeromycota, the phylum of arbuscular mycorrhizal fungi. Arch. Virol. 2014, 159, 2157–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezawa, T.; Ikeda, Y.; Shimura, H.; Masuta, C. Detection and characterization of mycoviruses in arbuscular mycorrhizal fungi by deep-sequencing. In Plant Virology Protocols; Humana Press: New York, NY, USA, 2015; pp. 171–180. [Google Scholar]
- Márquez, L.M.; Redman, R.S.; Rodriguez, R.J.; Roossinck, M.J. A virus in a fungus in a plant: Three-way symbiosis required for thermal tolerance. Science 2007, 315, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Turina, M.; Ghignone, S.; Astolfi, N.; Silvestri, A.; Bonfante, P.; Lanfranco, L. The virome of the arbuscular mycorrhizal fungus Gigaspora margarita reveals the first report of DNA fragments corresponding to replicating non-retroviral RNA viruses in fungi. Environ. Microbiol. 2018. [Google Scholar] [CrossRef]
- Pearson, M.N.; Beever, R.E.; Boine, B.; Arthur, K. Mycoviruses of filamentous fungi and their relevance to plant pathology. Mol. Plant Pathol. 2009, 10, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J. Evolutionary and ecological links between plant and fungal viruses. New Phytol. 2018, 221, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Hrabakova, L.; Koloniuk, I.; Petrzik, K. Phomopsis longicolla RNA virus 1—Novel virus at the edge of myco- and plant viruses. Virology 2017, 506, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. Origins and evolution of viruses of eukaryotes: The ultimate modularity. Virology 2015, 479–480, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Rastgou, M.; Habibi, M.K.; Izadpanah, K.; Masenga, V.; Milne, R.G.; Wolf, Y.I.; Koonin, E.V.; Turina, M. Molecular characterization of the plant virus genus Ourmiavirus and evidence of inter-kingdom reassortment of viral genome segments as its possible route of origin. J. Gen. Virol. 2009, 90, 2525–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nibert, M.L.; Vong, M.; Fugate, K.K.; Debat, H.J. Evidence for contemporary plant mitoviruses. Virology 2018, 518, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Bruenn, J.A.; Warner, B.E.; Yerramsetty, P. Widespread mitovirus sequences in plant genomes. PeerJ. 2015, 3, e876. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, D.K.; Jian, J.H.; Tavantzis, S.M. A double-stranded RNA element from a hypovirulent strain of Rhizoctonia solani occurs in DNA form and is genetically related to the pentafunctional AROM protein of the shikimate pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 6425–6429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzano, S.-Y.L.; Domier, L.L. Reprint of “Novel mycoviruses discovered from metatranscriptomics survey of soybean phyllosphere phytobiomes”. Virus Res. 2016, 219, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mensah, J.A.; Koch, A.M.; Antunes, P.M.; Kiers, E.T.; Hart, M.; Bucking, H. High functional diversity within species of arbuscular mycorrhizal fungi is associated with differences in phosphate and nitrogen uptake and fungal phosphate metabolism. Mycorrhiza 2015, 25, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Shimura, H.; Kitahara, R.; Masuta, C.; Ezawa, T. A novel virus-like double-stranded RNA in an obligate biotroph arbuscular mycorrhizal fungus: A hidden player in mycorrhizal symbiosis. Mol. Plant Microbe Interact. 2012, 25, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- St-Arnaud, M.; Hamel, C.; Vimard, B.; Caron, M.; Fortin, J. Enhanced hyphal growth and spore production of the arbuscular mycorrhizal fungus Glomus intraradices in an in vitro system in the absence of host roots. Mycol. Res. 1996, 100, 328–332. [Google Scholar] [CrossRef]
- Vierheilig, H.; Coughlan, A.P.; Wyss, U.; Piché, Y. Ink and vinegar, a simple staining technique for arbuscular-mycorrhizal fungi. Appl. Environ. Microbiol. 1998, 64, 5004–5007. [Google Scholar] [PubMed]
- McGonigle, T.P.; Miller, M.H.; Evans, D.G.; Fairchild, G.L.; Swan, J.A. A New Method which Gives an Objective Measure of Colonization of Roots by Vesicular-Arbuscular Mycorrhizal Fungi. New Phytol. 1990, 115, 495–501. [Google Scholar] [CrossRef]
- Tisserant, E.; Malbreil, M.; Kuo, A.; Kohler, A.; Symeonidi, A.; Balestrini, R.; Charron, P.; Duensing, N.; Frey, N.F.D.; Gianinazzi-Pearson, V.; et al. Genome of an arbuscular mycorrhizal fungus provides insight into the oldest plant symbiosis. Proc. Natl. Acad. Sci. USA 2013, 110, 20117–20122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüßler, A.; Walker, C. The Glomeromycota: A Species List with New Families and New Genera; Libraries at The Royal Botanic Garden Edinburgh, The Royal Botanic Garden Kew, Botanische Staatssammlung Munich, and Oregon State University: Kew, UK, 2010; pp. 1–56. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Pringle, F.M.; Zeddam, J.L.; Luke, B.T.; Cameron, C.E.; Kalmakoff, J.; Hanzlik, T.N.; Gordon, K.H.J.; Ward, V.K. The palm subdomain-based active site is internally permuted in viral RNA-dependent RNA polymerases of an ancient lineage. J. Mol. Biol. 2002, 324, 47–62. [Google Scholar] [CrossRef]
- Gohara, D.W.; Crotty, S.; Arnold, J.J.; Yoder, J.D.; Andino, R.; Cameron, C.E. Poliovirus RNA-dependent RNA polymerase (3Dpol): Structural, biochemical, and biological analysis of conserved structural motifs A and B. J. Biol. Chem. 2000, 275, 25523–25532. [Google Scholar] [CrossRef] [PubMed]
- Bartholomaus, A.; Wibberg, D.; Winkler, A.; Puhler, A.; Schluter, A.; Varrelmann, M. Deep Sequencing Analysis Reveals the Mycoviral Diversity of the Virome of an Avirulent Isolate of Rhizoctonia solani AG-2-2 IV. PLoS ONE 2016, 11, e0165965. [Google Scholar] [CrossRef]
- Fellbaum, C.R.; Mensah, J.A.; Cloos, A.J.; Strahan, G.E.; Pfeffer, P.E.; Kiers, E.T.; Bücking, H. Fungal nutrient allocation in common mycorrhizal networks is regulated by the carbon source strength of individual host plants. New Phytol. 2014, 203, 646–656. [Google Scholar] [CrossRef] [Green Version]
- Shackelton, L.A.; Holmes, E.C. The role of alternative genetic codes in viral evolution and emergence. J. Theor. Biol. 2008, 254, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Nibert, M.L. Mitovirus UGA (Trp) codon usage parallels that of host mitochondria. Virology 2017, 507, 96–100. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Hijikata, N.; Ohtomo, R.; Handa, Y.; Kawaguchi, M.; Saito, K.; Masuta, C.; Ezawa, T. Aquaporin-mediated long-distance polyphosphate translocation directed towards the host in arbuscular mycorrhizal symbiosis: Application of virus-induced gene silencing. New Phytol. 2016, 211, 1202–1208. [Google Scholar] [CrossRef]
- Otagaki, S.; Arai, M.; Takahashi, A.; Goto, K.; Hong, J.-S.; Masuta, C.; Kanazawa, A. Rapid induction of transcriptional and post-transcriptional gene silencing using a novel Cucumber mosaic virus vector. Plant Biotechnol. 2006, 23, 259–265. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contig Name | Data Source | Contig Length (nt) | Read Counts | NCBI Accession | Amino Acid Identity (%) | Putative Function (Most Similar Virus) |
|---|---|---|---|---|---|---|
| RdMV1 | SRX312982 (Rhizophagus diaphanum MUCL 43196) | 3554 | 1475 | MH732931 | 32 | RNA-dependent RNA polymerase [Rhizoctonia solani mitovirus 12] |
| RdMV2 | SRX312982 (Rhizophagus diaphanum MUCL 43196) | 4382 | 3649 | MH732930 | 28 | RNA-dependent RNA polymerase [Gigaspora margarita mitovirus 2] |
| RdMV3 | SRX312982 (Rhizophagus diaphanum MUCL 43196) | 3652 | 2350 | MK156099 | 36 | RNA-directed RNA polymerase [Rhizophagus sp. RF1 mitovirus] |
| RdMV4 | SRX312982 (Rhizophagus diaphanum MUCL 43196) | 3443 | 462 | MK156100 | 30 | RNA-dependent RNA polymerase [Rhizoctonia mitovirus 1] |
| RirMV1 | Sequenced transcriptome (submitted under accession: SRX4679168) | 3685 | 41,322 | MH732933 | 31 | RNA-dependent RNA polymerase [Rhizoctonia solani mitovirus 12] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neupane, A.; Feng, C.; Feng, J.; Kafle, A.; Bücking, H.; Lee Marzano, S.-Y. Metatranscriptomic Analysis and In Silico Approach Identified Mycoviruses in the Arbuscular Mycorrhizal Fungus Rhizophagus spp. Viruses 2018, 10, 707. https://doi.org/10.3390/v10120707
Neupane A, Feng C, Feng J, Kafle A, Bücking H, Lee Marzano S-Y. Metatranscriptomic Analysis and In Silico Approach Identified Mycoviruses in the Arbuscular Mycorrhizal Fungus Rhizophagus spp. Viruses. 2018; 10(12):707. https://doi.org/10.3390/v10120707
Chicago/Turabian StyleNeupane, Achal, Chenchen Feng, Jiuhuan Feng, Arjun Kafle, Heike Bücking, and Shin-Yi Lee Marzano. 2018. "Metatranscriptomic Analysis and In Silico Approach Identified Mycoviruses in the Arbuscular Mycorrhizal Fungus Rhizophagus spp." Viruses 10, no. 12: 707. https://doi.org/10.3390/v10120707
APA StyleNeupane, A., Feng, C., Feng, J., Kafle, A., Bücking, H., & Lee Marzano, S. -Y. (2018). Metatranscriptomic Analysis and In Silico Approach Identified Mycoviruses in the Arbuscular Mycorrhizal Fungus Rhizophagus spp. Viruses, 10(12), 707. https://doi.org/10.3390/v10120707