Modulation of Innate Immune Responses by the Influenza A NS1 and PA-X Proteins




Abstract
:1. Introduction
1.1. Influenza A Virus (IAV)’s Relevance to Human Health
1.2. Interferon and Inflammatory Cytokine Responses during IAV Infection
1.3. ISGs against IAV Infections
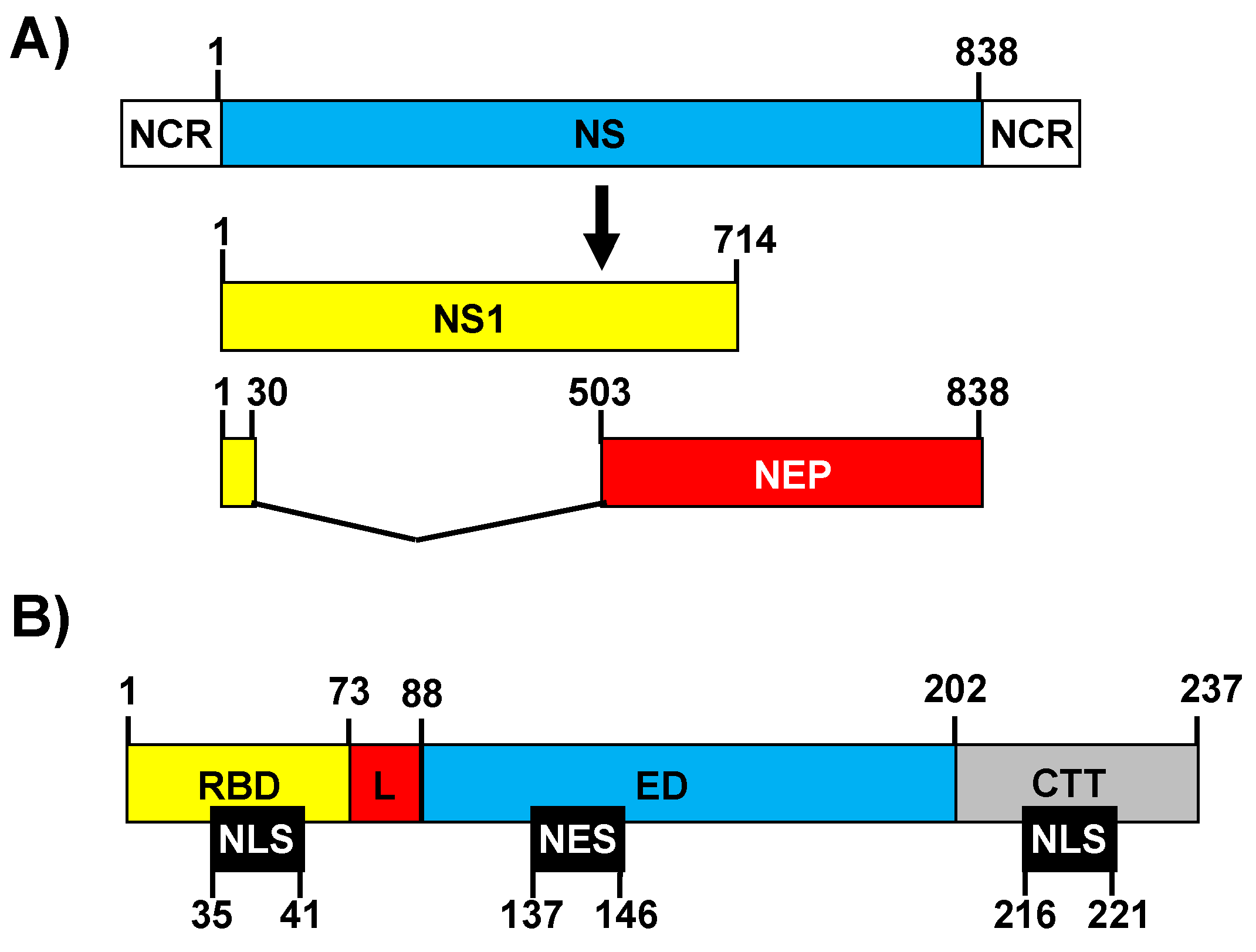
2. The IAV Non-Structural 1 (NS1) Protein
2.1. NS1 Protein Introduction
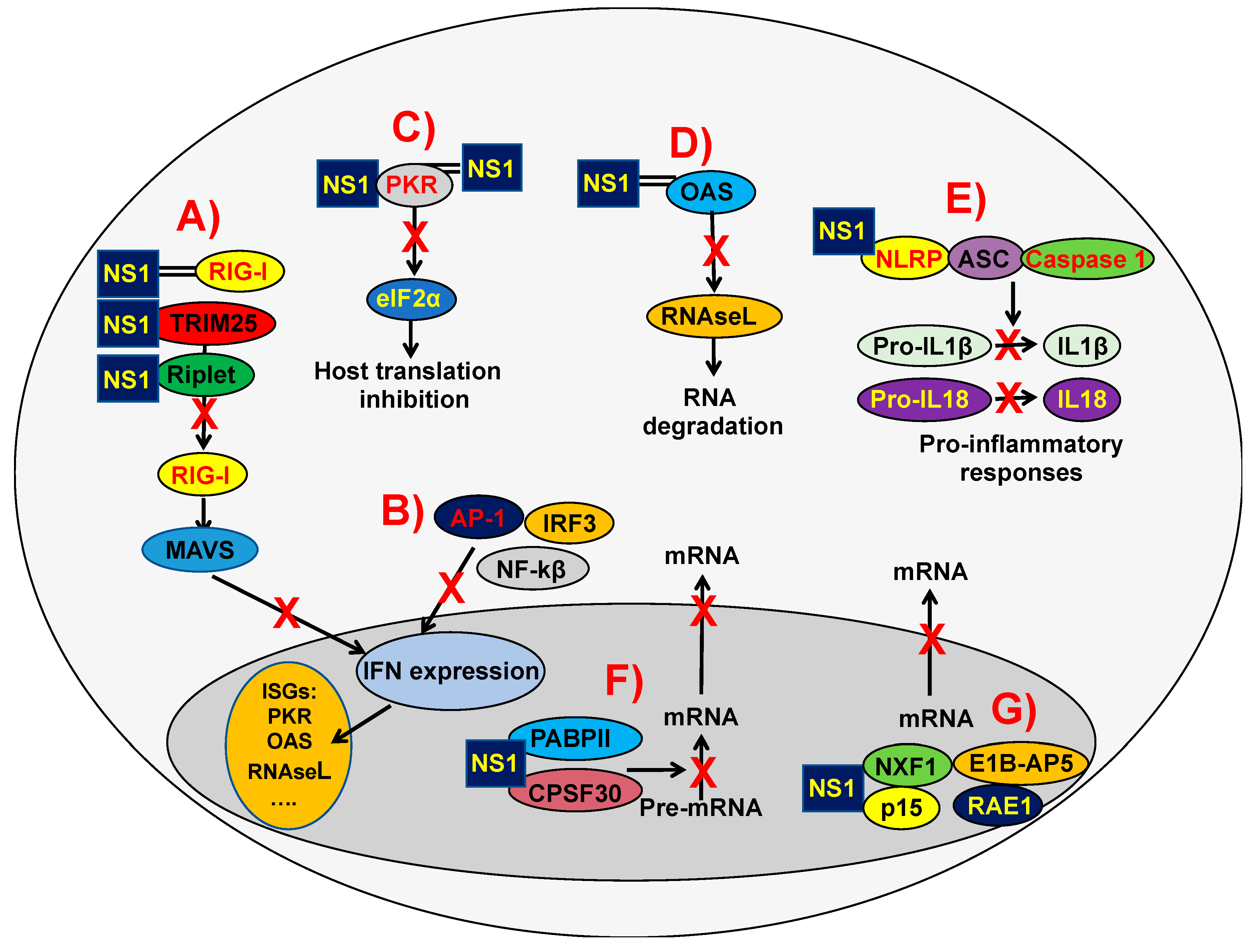
2.2. Mechanisms of IAV NS1 Protein Inhibition of Innate Immune Responses
2.3. Effect of the IAV NS1 Protein on Inhibition of Gene Expression
2.4. Effect of IAV NS1 on Virus Replication, Induction of Cytokines, and Pathogenesis
2.4.1. Effect of Partial and Complete NS1 Deletions on Virus Replication, Induction of Cytokines, and Pathogenesis
2.4.2. Effect of NS1 Mutations Affecting NS1-Mediated Host Shutoff on Innate Immune Responses and Virus Pathogenesis
2.4.3. Effect of NS1 Mutations Affecting Inflammatory Processes
2.5. Modulation of IAV NS1 Expression for the Development of More Efficient Live-Attenuated Influenza Vaccines (LAIV)
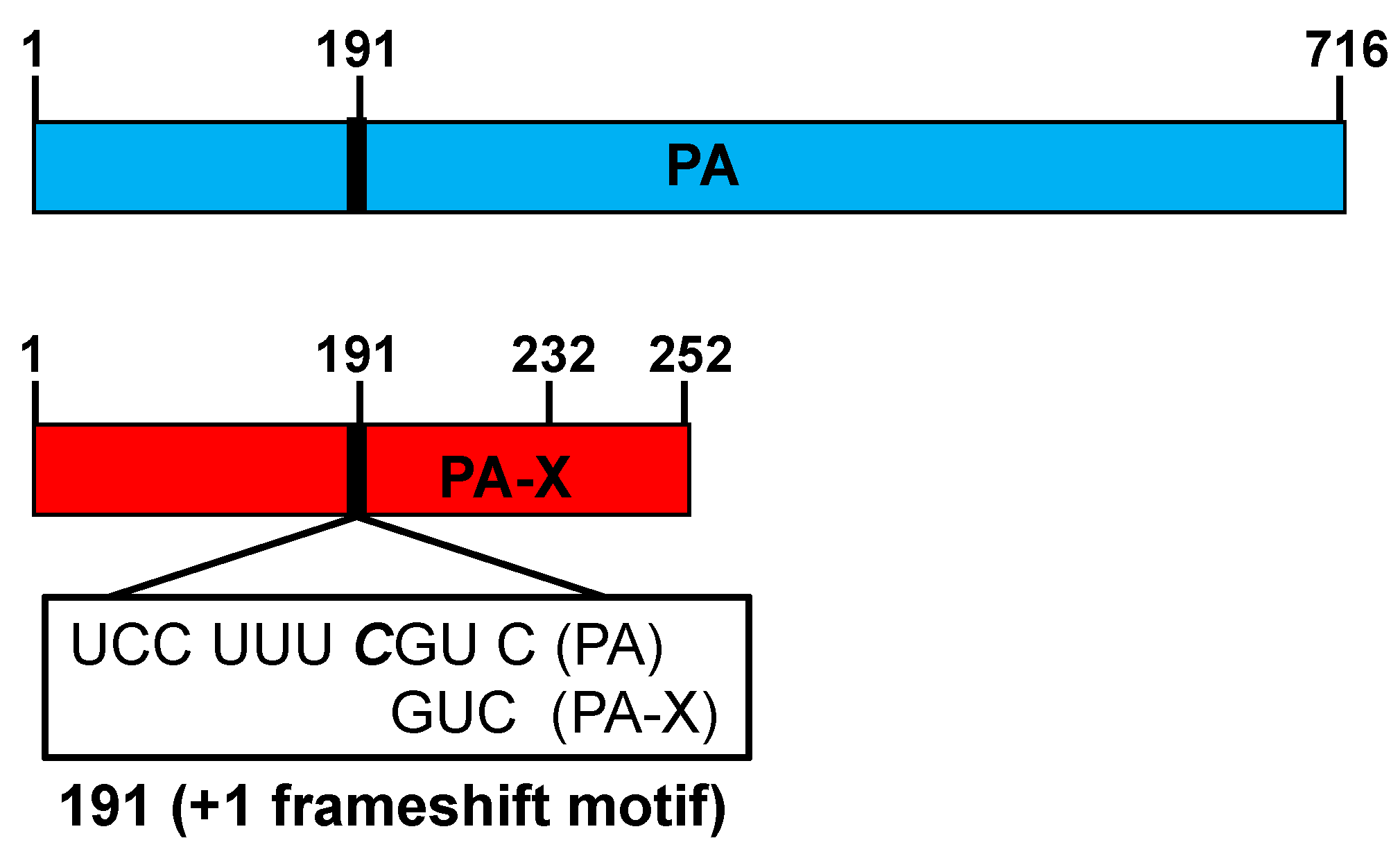
3. The IAV PA-X Protein
3.1. The IAV PA-X Protein’s Effect on Host Gene Expression
3.2. Effect of IAV PA-X on Virus Pathogenesis and Suppression of Innate Immune Responses
3.3. Modulation of PA-X to Generate LAIVs
4. Interplay of the IAV NS1 and PA-X Proteins
4.1. Interaction of the IAV NS1 and PA-X Proteins on Virus Fitness and Virulence
4.2. Inhibition of Host Gene Expression by IAV NS1 and PA-X Proteins for the Development of More Efficient LAIV
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACIP | Advisory Committee on Immunization Practices |
| ASC | Apoptosis-associated Speck-like containing a caspase-recruitment domain |
| ATF | Activating transcription factor |
| CD | Codon deoptimized |
| CIV | Canine influenza virus |
| CPSF30 | Cleavage and polyadenylation specificity factor 30 |
| CTT | C- terminal tail |
| dsRNA | double-stranded RNA |
| E1B-AP5 | Adenovirus early region 1B-associated protein 5 |
| ED | Effector domain |
| EIV | Equine influenza virus |
| eIF2α | α subunit of the eukaryotic initiation factor 2 |
| eIF4G | Eukaryotic initiation factor 4G |
| FDA | Food and drug administration |
| HA | Hemagglutinin |
| IAV | Influenza A virus |
| IFN | Interferon |
| IIV | Influenza inactivated vaccine |
| IKK | inhibitor of kappa β kinase |
| IL | Interleukin |
| IRF | Interferon regulatory factor |
| ISGs | Interferon (IFN)-stimulated genes |
| LAIV | Live-attenuated influenza vaccine |
| M2 | Matrix protein 2 |
| MDV | Master donor virus |
| NA | Neuraminidase |
| NCR | Non-coding region |
| NEP | Nuclear export protein |
| NES | Nuclear export signal |
| NF-κB | Nuclear factor kappa β |
| NLRP3 | NOD-like receptor family member LRR- and Pyrin domain containing-3 |
| NLS | Nuclear localization signal |
| NS1 | Non-structural protein 1 |
| NSs | NS split |
| NXF1 | Nuclear RNA export Factor 1 |
| OAS | Oligoadenylate synthase |
| ORF | Open reading frame |
| PA | Polymerase acid |
| PABP | Poly(A)-binding protein |
| PACT | Protein activator of the IFN-induced PKR |
| PAMPs | Pathogen-associated molecular patterns |
| PB1 | Polymerase basic 1 |
| PB2 | Polymerase basic 2 |
| pH1N1 | pandemic H1N1 virus |
| PI3K | p85-β of phosphatidylinositol 3-kinase |
| PKR | Protein kinase R |
| Pol II | Polymerase II |
| PR8 | A/Puerto Rico/8/34 |
| PRR | Pattern recognition receptor |
| RAE1 | Ribonucleic Acid Export 1 |
| RBD | RNA-binding domain |
| RIG-I | Retinoic acid inducible gene I |
| RNAse L | Ribonuclease L |
| RdRp | RNA-dependent RNA polymerase |
| ssRNA | single-stranded RNA |
| TNF | Tumor necrosis factor |
| TLRs | Toll-like receptors |
| TRIM25 | E3 ligase tripartite motif-containing protein 25 |
| UTR | Untranslated region |
| WT | Wild-type |
| WHO | World Health Organization |
| WSN | A/WSN/33 |
References
- Palese, P.; Shaw, M.L. Orthomyxoviridae: The viruses and their replication. In Fields Virology; Knipe, D., Howley, P., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006; pp. 1648–1689. [Google Scholar]
- Parrish, C.R.; Kawaoka, Y. The origins of new pandemic viruses: The acquisition of new host ranges by canine parvovirus and influenza A viruses. Annu. Rev. Microbiol. 2005, 59, 553–586. [Google Scholar] [CrossRef] [PubMed]
- Steel, J.; Lowen, A.C.; Mubareka, S.; Palese, P. Transmission of influenza virus in a mammalian host is increased by PB2 amino acids 627K or 627E/701N. PLoS Pathog. 2009, 5, e1000252. [Google Scholar] [CrossRef] [PubMed]
- Bailey, E.S.; Choi, J.Y.; Fieldhouse, J.K.; Borkenhagen, L.K.; Zemke, J.; Zhang, D.; Gray, G.C. The continual threat of influenza virus infections at the human-animal interface: What is new from a one health perspective? Evol. Med. Public Health 2018, 2018, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, G.; Marshall, J.F.; Morrell, J.; Robb, D.; McCauley, J.W.; Perez, D.R.; Parrish, C.R.; Murcia, P.R. Infection and pathogenesis of canine, equine, and human influenza viruses in canine tracheas. J. Virol. 2014, 88, 9208–9219. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.W.; Shay, D.K.; Weintraub, E.; Brammer, L.; Cox, N.; Anderson, L.J.; Fukuda, K. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 2003, 289, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Morens, D.M. Influenza: The once and future pandemic. Public Health Rep. 2010, 125 (Suppl. 3), 16–26. [Google Scholar] [CrossRef] [PubMed]
- Memorandum, W. A revision of the system of nomenclature for influenza viruses: A WHO memorandum. Bull. World Health Organ. 1980, 58, 585–591. [Google Scholar]
- Medina, R.A.; Garcia-Sastre, A. Influenza A viruses: New research developments. Nat. Rev. Microbiol. 2011, 9, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef]
- Kuchipudi, S.V.; Nissly, R.H. Novel flu viruses in bats and cattle: “Pushing the Envelope” of Influenza Infection. Vet. Sci. 2018, 5, 71. [Google Scholar] [CrossRef]
- Clark, A.M.; DeDiego, M.L.; Anderson, C.S.; Wang, J.; Yang, H.; Nogales, A.; Martinez-Sobrido, L.; Zand, M.S.; Sangster, M.Y.; Topham, D.J. Antigenicity of the 2015–2016 seasonal H1N1 human influenza virus HA and NA proteins. PLoS ONE 2017, 12, e0188267. [Google Scholar] [CrossRef] [PubMed]
- Grohskopf, L.A.; Sokolow, L.Z.; Broder, K.R.; Walter, E.B.; Bresee, J.S.; Fry, A.M.; Jernigan, D.B. Prevention and Control of Seasonal Influenza with Vaccines: Recommendations of the Advisory Committee on Immunization Practices-United States, 2017-18 Influenza Season. Am. J. Transplant. 2017, 17, 2970–2982. [Google Scholar] [CrossRef] [PubMed]
- Bui, C.M.; Chughtai, A.A.; Adam, D.C.; MacIntyre, C.R. An overview of the epidemiology and emergence of influenza A infection in humans over time. Arch. Public Health 2017, 75, 15. [Google Scholar] [CrossRef] [PubMed]
- Garten, R.J.; Davis, C.T.; Russell, C.A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W.M.; Xu, X.; Skepner, E.; Deyde, V.; et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 2009, 325, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, V.; Khiabanian, H.; Rabadan, R. Geographic dependence, surveillance, and origins of the 2009 influenza A (H1N1) virus. N. Engl. J. Med. 2009, 361, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.; Bahl, J.; Joseph, U.; Butt, K.M.; Peck, H.A.; Koay, E.S.; Oon, L.L.; Barr, I.G.; Vijaykrishna, D.; Smith, G.J. Phylodynamics of H1N1/2009 influenza reveals the transition from host adaptation to immune-driven selection. Nat. Commun. 2015, 6, 7952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.S.; Ortega, S.; Chaves, F.A.; Clark, A.M.; Yang, H.; Topham, D.J.; DeDiego, M.L. Natural and directed antigenic drift of the H1 influenza virus hemagglutinin stalk domain. Sci. Rep. 2017, 7, 14614. [Google Scholar] [CrossRef]
- Girard, M.P.; Cherian, T.; Pervikov, Y.; Kieny, M.P. A review of vaccine research and development: Human acute respiratory infections. Vaccine 2005, 23, 5708–5724. [Google Scholar] [CrossRef]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Goff, P.H.; Hayashi, T.; He, W.; Yao, S.; Cottam, H.B.; Tan, G.S.; Crain, B.; Krammer, F.; Messer, K.; Pu, M.; et al. Synthetic Toll-Like Receptor 4 (TLR4) and TLR7 Ligands Work Additively via MyD88 To Induce Protective Antiviral Immunity in Mice. J. Virol. 2017, 91, JVI-01050. [Google Scholar] [CrossRef]
- Weber-Gerlach, M.; Weber, F. Standing on three legs: Antiviral activities of RIG-I against influenza viruses. Curr. Opin. Immunol. 2016, 42, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 2014, 516, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Wolff, T.; Ludwig, S. Influenza viruses control the vertebrate type I interferon system: Factors, mechanisms, and consequences. J. Interferon Cytokine Res. 2009, 29, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Hermant, P.; Michiels, T. Interferon-lambda in the context of viral infections: Production, response and therapeutic implications. J. Innate Immun. 2014, 6, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H.; Paludan, S.R. Molecular pathways in virus-induced cytokine production. Microbiol. Mol. Biol. Rev. 2001, 65, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Crotta, S.; Davidson, S.; Mahlakoiv, T.; Desmet, C.J.; Buckwalter, M.R.; Albert, M.L.; Staeheli, P.; Wack, A. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog. 2013, 9, e1003773. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Marie, I.J.; Durbin, J.E. Induction and function of type I and III interferon in response to viral infection. Curr. Opin. Virol. 2011, 1, 476–486. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, H.H.; Schneider, W.M.; Rice, C.M. Interferons and viruses: An evolutionary arms race of molecular interactions. Trends Immunol. 2015, 36, 124–138. [Google Scholar] [CrossRef]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT signaling: From interferons to cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef]
- Tenoever, B.R.; Ng, S.L.; Chua, M.A.; McWhirter, S.M.; Garcia-Sastre, A.; Maniatis, T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science 2007, 315, 1274–1278. [Google Scholar] [CrossRef]
- Ng, S.L.; Friedman, B.A.; Schmid, S.; Gertz, J.; Myers, R.M.; tenOever, B.R.; Maniatis, T. I kappa B kinase epsilon (IKK epsilon) regulates the balance between type I and type II interferon responses. Proc. Natl. Acad. Sci. USA 2011, 108, 21170–21175. [Google Scholar] [CrossRef] [PubMed]
- Groslambert, M.; Py, B.F. Spotlight on the NLRP3 inflammasome pathway. J. Inflamm. Res. 2018, 11, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Guarda, G.; Zenger, M.; Yazdi, A.S.; Schroder, K.; Ferrero, I.; Menu, P.; Tardivel, A.; Mattmann, C.; Tschopp, J. Differential expression of NLRP3 among hematopoietic cells. J. Immunol. 2011, 186, 2529–2534. [Google Scholar] [CrossRef] [PubMed]
- Pothlichet, J.; Meunier, I.; Davis, B.K.; Ting, J.P.; Skamene, E.; von Messling, V.; Vidal, S.M. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog. 2013, 9, e1003256. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAuley, J.L.; Tate, M.D.; MacKenzie-Kludas, C.J.; Pinar, A.; Zeng, W.; Stutz, A.; Latz, E.; Brown, L.E.; Mansell, A. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 2013, 9, e1003392. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Villalon-Letelier, F.; Brooks, A.G.; Saunders, P.M.; Londrigan, S.L.; Reading, P.C. Host cell restriction factors that limit influenza a infection. Viruses 2017, 9, 376. [Google Scholar] [CrossRef]
- Staeheli, P.; Haller, O.; Boll, W.; Lindenmann, J.; Weissmann, C. Mx protein: Constitutive expression in 3T3 cells transformed with cloned Mx cDNA confers selective resistance to influenza virus. Cell 1986, 44, 147–158. [Google Scholar] [CrossRef]
- Krug, R.M.; Shaw, M.; Broni, B.; Shapiro, G.; Haller, O. Inhibition of influenza viral mRNA synthesis in cells expressing the interferon-induced Mx gene product. J. Virol. 1985, 56, 201–206. [Google Scholar] [PubMed]
- Haller, O.; Arnheiter, H.; Pavlovic, J.; Staeheli, P. The Discovery of the Antiviral Resistance Gene Mx: A Story of Great Ideas, Great Failures, and Some Success. Annu. Rev. Virol. 2018, 5, 33–51. [Google Scholar] [CrossRef] [PubMed]
- Turan, K.; Mibayashi, M.; Sugiyama, K.; Saito, S.; Numajiri, A.; Nagata, K. Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic Acids Res. 2004, 32, 643–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlovic, J.; Arzet, H.A.; Hefti, H.P.; Frese, M.; Rost, D.; Ernst, B.; Kolb, E.; Staeheli, P.; Haller, O. Enhanced virus resistance of transgenic mice expressing the human MxA protein. J. Virol. 1995, 69, 4506–4510. [Google Scholar] [PubMed]
- Goujon, C.; Moncorge, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hue, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.C.; Zhong, G.; Huang, I.C.; Farzan, M. IFITM-Family Proteins: The Cell’s First Line of Antiviral Defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Clemens, M.J.; Williams, B.R. Inhibition of cell-free protein synthesis by pppA2′p5′A2′p5′A: A novel oligonucleotide synthesized by interferon-treated L cell extracts. Cell 1978, 13, 565–572. [Google Scholar] [CrossRef]
- Silverman, R.H.; Weiss, S.R. Viral phosphodiesterases that antagonize double-stranded RNA signaling to RNase L by degrading 2-5A. J. Interferon Cytokine Res. 2014, 34, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Malathi, K.; Dong, B.; Gale, M., Jr.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drappier, M.; Michiels, T. Inhibition of the OAS/RNase L pathway by viruses. Curr. Opin. Virol. 2015, 15, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.C.; Sen, G.C. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998, 17, 4379–4390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Haque, J.; Lacoste, J.; Hiscott, J.; Williams, B.R. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc. Natl. Acad. Sci. USA 1994, 91, 6288–6292. [Google Scholar] [CrossRef] [PubMed]
- Schulz, O.; Pichlmair, A.; Rehwinkel, J.; Rogers, N.C.; Scheuner, D.; Kato, H.; Takeuchi, O.; Akira, S.; Kaufman, R.J.; Reis e Sousa, C. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe 2010, 7, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Roberts, P.C.; Brown, L.E.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000, 13, 129–141. [Google Scholar] [CrossRef]
- Lamb, R.A.; Lai, C.J. Sequence of interrupted and uninterrupted mRNAs and cloned DNA coding for the two overlapping nonstructural proteins of influenza virus. Cell 1980, 21, 475–485. [Google Scholar] [CrossRef]
- Robb, N.C.; Jackson, D.; Vreede, F.T.; Fodor, E. Splicing of influenza A virus NS1 mRNA is independent of the viral NS1 protein. J. Gen. Virol. 2010, 91, 2331–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marc, D. Influenza virus non-structural protein NS1: Interferon antagonism and beyond. J. Gen. Virol. 2014, 95, 2594–2611. [Google Scholar] [CrossRef]
- Lohrmann, F.; Dijkman, R.; Stertz, S.; Thiel, V.; Haller, O.; Staeheli, P.; Kochs, G. Emergence of a C-terminal seven-amino-acid elongation of NS1 in around 1950 conferred a minor growth advantage to former seasonal influenza A viruses. J. Virol. 2013, 87, 11300–11303. [Google Scholar] [CrossRef]
- Chauche, C.; Nogales, A.; Zhu, H.; Goldfarb, D.; Ahmad Shanizza, A.I.; Gu, Q.; Parrish, C.R.; Martinez-Sobrido, L.; Marshall, J.F.; Murcia, P.R. Mammalian adaptation of an avian influenza A virus involves stepwise changes in NS1. J. Virol. 2018, 92, JVI-01875. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.Y.; Tejero, R.; Huang, Y.; Zimmerman, D.E.; Rios, C.B.; Krug, R.M.; Montelione, G.T. A novel RNA-binding motif in influenza A virus non-structural protein 1. Nat. Struct. Biol. 1997, 4, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lynch, P.A.; Chien, C.Y.; Montelione, G.T.; Krug, R.M.; Berman, H.M. Crystal structure of the unique RNA-binding domain of the influenza virus NS1 protein. Nat. Struct. Biol. 1997, 4, 896–899. [Google Scholar] [CrossRef]
- Wang, W.; Riedel, K.; Lynch, P.; Chien, C.Y.; Montelione, G.T.; Krug, R.M. RNA binding by the novel helical domain of the influenza virus NS1 protein requires its dimer structure and a small number of specific basic amino acids. RNA 1999, 5, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donelan, N.R.; Basler, C.F.; Garcia-Sastre, A. A recombinant influenza A virus expressing an RNA-binding-defective NS1 protein induces high levels of beta interferon and is attenuated in mice. J. Virol. 2003, 77, 13257–13266. [Google Scholar] [CrossRef] [PubMed]
- Greenspan, D.; Palese, P.; Krystal, M. Two nuclear location signals in the influenza virus NS1 nonstructural protein. J. Virol. 1988, 62, 3020–3026. [Google Scholar] [PubMed]
- Melen, K.; Kinnunen, L.; Fagerlund, R.; Ikonen, N.; Twu, K.Y.; Krug, R.M.; Julkunen, I. Nuclear and nucleolar targeting of influenza A virus NS1 protein: Striking differences between different virus subtypes. J. Virol. 2007, 81, 5995–6006. [Google Scholar] [CrossRef]
- Li, Y.; Yamakita, Y.; Krug, R.M. Regulation of a nuclear export signal by an adjacent inhibitory sequence: The effector domain of the influenza virus NS1 protein. Proc. Natl. Acad. Sci. USA 1998, 95, 4864–4869. [Google Scholar] [CrossRef] [Green Version]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef] [Green Version]
- Nogales, A.; Martinez-Sobrido, L.; Chiem, K.; Topham, D.J.; DeDiego, M.L. Functional Evolution of the 2009 Pandemic H1N1 Influenza NS1 and PA in Humans. J. Virol. 2018, 92, e01206-18. [Google Scholar] [CrossRef]
- Nogales, A.; Chauche, C.; DeDiego, M.L.; Topham, D.J.; Parrish, C.R.; Murcia, P.R.; Martinez-Sobrido, L. The K186E Amino Acid Substitution in the Canine Influenza Virus H3N8 NS1 Protein Restores Its Ability to Inhibit Host Gene Expression. J. Virol. 2017, 91, JVI-00877. [Google Scholar] [CrossRef] [PubMed]
- Steidle, S.; Martinez-Sobrido, L.; Mordstein, M.; Lienenklaus, S.; Garcia-Sastre, A.; Staheli, P.; Kochs, G. Glycine 184 in nonstructural protein NS1 determines the virulence of influenza A virus strain PR8 without affecting the host interferon response. J. Virol. 2010, 84, 12761–12770. [Google Scholar] [CrossRef] [PubMed]
- Kochs, G.; Garcia-Sastre, A.; Martinez-Sobrido, L. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 2007, 81, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G. Conformational plasticity of the influenza A virus NS1 protein. J. Gen. Virol. 2014, 95, 2099–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, D.; Hossain, M.J.; Hickman, D.; Perez, D.R.; Lamb, R.A. A new influenza virus virulence determinant: The NS1 protein four C-terminal residues modulate pathogenicity. Proc. Natl. Acad. Sci. USA 2008, 105, 4381–4386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemm, C.; Boergeling, Y.; Ludwig, S.; Ehrhardt, C. Immunomodulatory nonstructural proteins of influenza a Viruses. Trends Microbiol. 2018, 26, 624–636. [Google Scholar] [CrossRef]
- Garcia-Sastre, A.; Egorov, A.; Matassov, D.; Brandt, S.; Levy, D.E.; Durbin, J.E.; Palese, P.; Muster, T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 1998, 252, 324–330. [Google Scholar] [CrossRef]
- Kochs, G.; Koerner, I.; Thiel, L.; Kothlow, S.; Kaspers, B.; Ruggli, N.; Summerfield, A.; Pavlovic, J.; Stech, J.; Staeheli, P. Properties of H7N7 influenza A virus strain SC35M lacking interferon antagonist NS1 in mice and chickens. J. Gen. Virol. 2007, 88, 1403–1409. [Google Scholar] [CrossRef] [Green Version]
- Nogales, A.; Huang, K.; Chauche, C.; DeDiego, M.L.; Murcia, P.R.; Parrish, C.R.; Martinez-Sobrido, L. Canine influenza viruses with modified NS1 proteins for the development of live-attenuated vaccines. Virology 2017, 500, 1–10. [Google Scholar] [CrossRef]
- Talon, J.; Salvatore, M.; O’Neill, R.E.; Nakaya, Y.; Zheng, H.; Muster, T.; Garcia-Sastre, A.; Palese, P. Influenza A and B viruses expressing altered NS1 proteins: A vaccine approach. Proc. Natl. Acad. Sci. USA 2000, 97, 4309–4314. [Google Scholar] [CrossRef] [Green Version]
- Nogales, A.; Baker, S.F.; Ortiz-Riano, E.; Dewhurst, S.; Topham, D.J.; Martinez-Sobrido, L. Influenza A virus attenuation by codon deoptimization of the NS gene for vaccine development. J. Virol. 2014, 88, 10525–10540. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Nogales, A.; Lambert-Emo, K.; Martinez-Sobrido, L.; Topham, D.J. NS1 Protein Mutation I64T Affects Interferon Responses and Virulence of Circulating H3N2 Human Influenza A Viruses. J. Virol. 2016, 90, 9693–9711. [Google Scholar] [CrossRef]
- Nogales, A.; Martinez-Sobrido, L.; Topham, D.J.; DeDiego, M.L. NS1 Protein Amino Acid Changes D189N and V194I Affect Interferon Responses, Thermosensitivity, and Virulence of Circulating H3N2 Human Influenza A Viruses. J. Virol. 2017, 91, e01930-16. [Google Scholar] [CrossRef]
- Trapp, S.; Soubieux, D.; Lidove, A.; Esnault, E.; Lion, A.; Guillory, V.; Wacquiez, A.; Kut, E.; Quere, P.; Larcher, T.; et al. Major contribution of the RNA-binding domain of NS1 in the pathogenicity and replication potential of an avian H7N1 influenza virus in chickens. Virol. J. 2018, 15, 55. [Google Scholar] [CrossRef] [Green Version]
- Newby, C.M.; Sabin, L.; Pekosz, A. The RNA binding domain of influenza A virus NS1 protein affects secretion of tumor necrosis factor alpha, interleukin-6, and interferon in primary murine tracheal epithelial cells. J. Virol. 2007, 81, 9469–9480. [Google Scholar] [CrossRef]
- Min, J.Y.; Krug, R.M. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2’-5’ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 7100–7105. [Google Scholar] [CrossRef]
- Mibayashi, M.; Martinez-Sobrido, L.; Loo, Y.M.; Cardenas, W.B.; Gale, M., Jr.; Garcia-Sastre, A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef]
- Guo, Z.; Chen, L.M.; Zeng, H.; Gomez, J.A.; Plowden, J.; Fujita, T.; Katz, J.M.; Donis, R.O.; Sambhara, S. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 2007, 36, 263–269. [Google Scholar] [CrossRef]
- Opitz, B.; Rejaibi, A.; Dauber, B.; Eckhard, J.; Vinzing, M.; Schmeck, B.; Hippenstiel, S.; Suttorp, N.; Wolff, T. IFNbeta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell. Microbiol. 2007, 9, 930–938. [Google Scholar] [CrossRef]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; Garcia-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef]
- Rajsbaum, R.; Albrecht, R.A.; Wang, M.K.; Maharaj, N.P.; Versteeg, G.A.; Nistal-Villan, E.; Garcia-Sastre, A.; Gack, M.U. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 2012, 8, e1003059. [Google Scholar] [CrossRef]
- Koliopoulos, M.G.; Lethier, M.; van der Veen, A.G.; Haubrich, K.; Hennig, J.; Kowalinski, E.; Stevens, R.V.; Martin, S.R.; Reis e Sousa, C.; Cusack, S.; et al. Molecular mechanism of influenza A NS1-mediated TRIM25 recognition and inhibition. Nat. Commun. 2018, 9, 1820. [Google Scholar] [CrossRef]
- Feng, W.; Sun, X.; Shi, N.; Zhang, M.; Guan, Z.; Duan, M. Influenza a virus NS1 protein induced A20 contributes to viral replication by suppressing interferon-induced antiviral response. Biochem. Biophys. Res. Commun. 2017, 482, 1107–1113. [Google Scholar] [CrossRef]
- Maelfait, J.; Roose, K.; Vereecke, L.; Mc Guire, C.; Sze, M.; Schuijs, M.J.; Willart, M.; Ibanez, L.I.; Hammad, H.; Lambrecht, B.N.; et al. A20 Deficiency in Lung Epithelial Cells Protects against Influenza A Virus Infection. PLoS Pathog. 2016, 12, e1005410. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef]
- Marazzi, I.; Ho, J.S.; Kim, J.; Manicassamy, B.; Dewell, S.; Albrecht, R.A.; Seibert, C.W.; Schaefer, U.; Jeffrey, K.L.; Prinjha, R.K.; et al. Suppression of the antiviral response by an influenza histone mimic. Nature 2012, 483, 428–433. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Song, L.; Li, J.; Zhang, Z.; Peng, H.; Jiang, W.; Wang, Q.; Kang, T.; Chen, S.; Huang, W. Influenza A virus-encoded NS1 virulence factor protein inhibits innate immune response by targeting IKK. Cell. Microbiol. 2012, 14, 1849–1866. [Google Scholar] [CrossRef] [Green Version]
- Talon, J.; Horvath, C.M.; Polley, R.; Basler, C.F.; Muster, T.; Palese, P.; Garcia-Sastre, A. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J. Virol. 2000, 74, 7989–7996. [Google Scholar] [CrossRef]
- Ludwig, S.; Wang, X.; Ehrhardt, C.; Zheng, H.; Donelan, N.; Planz, O.; Pleschka, S.; Garcia-Sastre, A.; Heins, G.; Wolff, T. The influenza A virus NS1 protein inhibits activation of Jun N-terminal kinase and AP-1 transcription factors. J. Virol. 2002, 76, 11166–11171. [Google Scholar] [CrossRef]
- Li, S.; Min, J.Y.; Krug, R.M.; Sen, G.C. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 2006, 349, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Min, J.Y.; Li, S.; Sen, G.C.; Krug, R.M. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology 2007, 363, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Schierhorn, K.L.; Jolmes, F.; Bespalowa, J.; Saenger, S.; Peteranderl, C.; Dzieciolowski, J.; Mielke, M.; Budt, M.; Pleschka, S.; Herrmann, A.; et al. Influenza A Virus Virulence Depends on Two Amino Acids in the N-Terminal Domain of Its NS1 Protein To Facilitate Inhibition of the RNA-Dependent Protein Kinase PKR. J. Virol. 2017, 91, e00198-17. [Google Scholar] [CrossRef]
- Tawaratsumida, K.; Phan, V.; Hrincius, E.R.; High, A.A.; Webby, R.; Redecke, V.; Hacker, H. Quantitative proteomic analysis of the influenza A virus nonstructural proteins NS1 and NS2 during natural cell infection identifies PACT as an NS1 target protein and antiviral host factor. J. Virol. 2014, 88, 9038–9048. [Google Scholar] [CrossRef]
- Cheong, W.C.; Kang, H.R.; Yoon, H.; Kang, S.J.; Ting, J.P.; Song, M.J. Influenza A Virus NS1 Protein Inhibits the NLRP3 Inflammasome. PLoS ONE 2015, 10, e0126456. [Google Scholar] [CrossRef]
- Park, H.S.; Liu, G.; Thulasi Raman, S.N.; Landreth, S.L.; Liu, Q.; Zhou, Y. NS1 Protein of 2009 Pandemic Influenza A Virus Inhibits Porcine NLRP3 Inflammasome-Mediated Interleukin-1 Beta Production by Suppressing ASC Ubiquitination. J. Virol. 2018, 92, JVI-00022. [Google Scholar] [CrossRef]
- Moriyama, M.; Chen, I.Y.; Kawaguchi, A.; Koshiba, T.; Nagata, K.; Takeyama, H.; Hasegawa, H.; Ichinohe, T. The RNA- and TRIM25-Binding Domains of Influenza Virus NS1 Protein Are Essential for Suppression of NLRP3 Inflammasome-Mediated Interleukin-1beta Secretion. J. Virol. 2016, 90, 4105–4114. [Google Scholar] [CrossRef]
- Das, K.; Ma, L.C.; Xiao, R.; Radvansky, B.; Aramini, J.; Zhao, L.; Marklund, J.; Kuo, R.L.; Twu, K.Y.; Arnold, E.; et al. Structural basis for suppression of a host antiviral response by influenza A virus. Proc. Natl. Acad. Sci. USA 2008, 105, 13093–13098. [Google Scholar] [CrossRef] [Green Version]
- Noah, D.L.; Twu, K.Y.; Krug, R.M. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3′ end processing of cellular pre-mRNAS. Virology 2003, 307, 386–395. [Google Scholar] [CrossRef]
- Nemeroff, M.E.; Barabino, S.M.; Li, Y.; Keller, W.; Krug, R.M. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3’end formation of cellular pre-mRNAs. Mol. Cell 1998, 1, 991–1000. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Krug, R.M. Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3’-end processing machinery. EMBO J. 1999, 18, 2273–2283. [Google Scholar] [CrossRef]
- Tu, J.; Guo, J.; Zhang, A.; Zhang, W.; Zhao, Z.; Zhou, H.; Liu, C.; Chen, H.; Jin, M. Effects of the C-terminal truncation in NS1 protein of the 2009 pandemic H1N1 influenza virus on host gene expression. PLoS ONE 2011, 6, e26175. [Google Scholar] [CrossRef]
- Aragon, T.; de la Luna, S.; Novoa, I.; Carrasco, L.; Ortin, J.; Nieto, A. Eukaryotic translation initiation factor 4GI is a cellular target for NS1 protein, a translational activator of influenza virus. Mol. Cell. Biol. 2000, 20, 6259–6268. [Google Scholar] [CrossRef]
- Hale, B.G.; Jackson, D.; Chen, Y.H.; Lamb, R.A.; Randall, R.E. Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Anderson, D.H.; Liu, Q.; Zhou, Y. Mechanism of influenza A virus NS1 protein interaction with the p85beta, but not the p85alpha, subunit of phosphatidylinositol 3-kinase (PI3K) and up-regulation of PI3K activity. J. Biol. Chem. 2008, 283, 23397–23409. [Google Scholar] [CrossRef]
- Satterly, N.; Tsai, P.L.; van Deursen, J.; Nussenzveig, D.R.; Wang, Y.; Faria, P.A.; Levay, A.; Levy, D.E.; Fontoura, B.M. Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2007, 104, 1853–1858. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, M.; Zheng, H.; Muster, T.; Palese, P.; Beg, A.A.; Garcia-Sastre, A. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J. Virol. 2000, 74, 11566–11573. [Google Scholar] [CrossRef]
- Stasakova, J.; Ferko, B.; Kittel, C.; Sereinig, S.; Romanova, J.; Katinger, H.; Egorov, A. Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1beta and 18. J. Gen. Virol. 2005, 86, 185–195. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Wolff, T.; Pleschka, S.; Planz, O.; Beermann, W.; Bode, J.G.; Schmolke, M.; Ludwig, S. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J. Virol. 2007, 81, 3058–3067. [Google Scholar] [CrossRef]
- Hrincius, E.R.; Dierkes, R.; Anhlan, D.; Wixler, V.; Ludwig, S.; Ehrhardt, C. Phosphatidylinositol-3-kinase (PI3K) is activated by influenza virus vRNA via the pathogen pattern receptor Rig-I to promote efficient type I interferon production. Cell. Microbiol. 2011, 13, 1907–1919. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Masic, A.; Liu, Q.; Zhou, Y. Regulation of influenza A virus induced CXCL-10 gene expression requires PI3K/Akt pathway and IRF3 transcription factor. Mol. Immunol. 2011, 48, 1417–1423. [Google Scholar] [CrossRef]
- Shin, Y.K.; Li, Y.; Liu, Q.; Anderson, D.H.; Babiuk, L.A.; Zhou, Y. SH3 binding motif 1 in influenza A virus NS1 protein is essential for PI3K/Akt signaling pathway activation. J. Virol. 2007, 81, 12730–12739. [Google Scholar] [CrossRef]
- Chan, S.; Choi, E.A.; Shi, Y. Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip. Rev. RNA 2011, 2, 321–335. [Google Scholar] [CrossRef]
- Levene, R.E.; Gaglia, M.M. Host Shutoff in Influenza A Virus: Many Means to an End. Viruses 2018, 10, 475. [Google Scholar] [CrossRef]
- Hale, B.G.; Steel, J.; Medina, R.A.; Manicassamy, B.; Ye, J.; Hickman, D.; Hai, R.; Schmolke, M.; Lowen, A.C.; Perez, D.R.; et al. Inefficient control of host gene expression by the 2009 pandemic H1N1 influenza A virus NS1 protein. J. Virol. 2010, 84, 6909–6922. [Google Scholar] [CrossRef]
- Clark, A.M.; Nogales, A.; Martinez-Sobrido, L.; Topham, D.J.; DeDiego, M.L. Functional Evolution of Influenza Virus NS1 Protein in Currently Circulating Human 2009 Pandemic H1N1 Viruses. J. Virol. 2017, 91, JVI-00721. [Google Scholar] [CrossRef]
- Hale, B.G.; Steel, J.; Manicassamy, B.; Medina, R.A.; Ye, J.; Hickman, D.; Lowen, A.C.; Perez, D.R.; Garcia-Sastre, A. Mutations in the NS1 C-terminal tail do not enhance replication or virulence of the 2009 pandemic H1N1 influenza A virus. J. Gen. Virol. 2010, 91, 1737–1742. [Google Scholar] [CrossRef] [Green Version]
- Anastasina, M.; Le May, N.; Bugai, A.; Fu, Y.; Soderholm, S.; Gaelings, L.; Ohman, T.; Tynell, J.; Kyttanen, S.; Barboric, M.; et al. Influenza virus NS1 protein binds cellular DNA to block transcription of antiviral genes. Biochim. Biophys. Acta 2016, 1859, 1440–1448. [Google Scholar] [CrossRef] [Green Version]
- Pica, N.; Langlois, R.A.; Krammer, F.; Margine, I.; Palese, P. NS1-truncated live attenuated virus vaccine provides robust protection to aged mice from viral challenge. J. Virol. 2012, 86, 10293–10301. [Google Scholar] [CrossRef]
- Kochs, G.; Martinez-Sobrido, L.; Lienenklaus, S.; Weiss, S.; Garcia-Sastre, A.; Staeheli, P. Strong interferon-inducing capacity of a highly virulent variant of influenza A virus strain PR8 with deletions in the NS1 gene. J. Gen. Virol. 2009, 90, 2990–2994. [Google Scholar] [CrossRef] [Green Version]
- Steel, J.; Lowen, A.C.; Pena, L.; Angel, M.; Solorzano, A.; Albrecht, R.; Perez, D.R.; Garcia-Sastre, A.; Palese, P. Live attenuated influenza viruses containing NS1 truncations as vaccine candidates against H5N1 highly pathogenic avian influenza. J. Virol. 2009, 83, 1742–1753. [Google Scholar] [CrossRef]
- Baskin, C.R.; Bielefeldt-Ohmann, H.; Garcia-Sastre, A.; Tumpey, T.M.; Van Hoeven, N.; Carter, V.S.; Thomas, M.J.; Proll, S.; Solorzano, A.; Billharz, R.; et al. Functional genomic and serological analysis of the protective immune response resulting from vaccination of macaques with an NS1-truncated influenza virus. J. Virol. 2007, 81, 11817–11827. [Google Scholar] [CrossRef]
- Chambers, T.M.; Quinlivan, M.; Sturgill, T.; Cullinane, A.; Horohov, D.W.; Zamarin, D.; Arkins, S.; Garcia-Sastre, A.; Palese, P. Influenza A viruses with truncated NS1 as modified live virus vaccines: Pilot studies of safety and efficacy in horses. Equine Vet. J. 2009, 41, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Quinlivan, M.; Zamarin, D.; Garcia-Sastre, A.; Cullinane, A.; Chambers, T.; Palese, P. Attenuation of equine influenza viruses through truncations of the NS1 protein. J. Virol. 2005, 79, 8431–8439. [Google Scholar] [CrossRef]
- Ayllon, J.; Domingues, P.; Rajsbaum, R.; Miorin, L.; Schmolke, M.; Hale, B.G.; Garcia-Sastre, A. A single amino acid substitution in the novel H7N9 influenza A virus NS1 protein increases CPSF30 binding and virulence. J. Virol. 2014, 88, 12146–12151. [Google Scholar] [CrossRef]
- Spesock, A.; Malur, M.; Hossain, M.J.; Chen, L.M.; Njaa, B.L.; Davis, C.T.; Lipatov, A.S.; York, I.A.; Krug, R.M.; Donis, R.O. The virulence of 1997 H5N1 influenza viruses in the mouse model is increased by correcting a defect in their NS1 proteins. J. Virol. 2011, 85, 7048–7058. [Google Scholar] [CrossRef]
- Kuo, R.L.; Zhao, C.; Malur, M.; Krug, R.M. Influenza A virus strains that circulate in humans differ in the ability of their NS1 proteins to block the activation of IRF3 and interferon-beta transcription. Virology 2010, 408, 146–158. [Google Scholar] [CrossRef]
- Twu, K.Y.; Kuo, R.L.; Marklund, J.; Krug, R.M. The H5N1 influenza virus NS genes selected after 1998 enhance virus replication in mammalian cells. J. Virol. 2007, 81, 8112–8121. [Google Scholar] [CrossRef]
- Kaewborisuth, C.; Kaplan, B.; Zanin, M.; Finkelstein, D.; Webby, R.J.; Lekcharoensuk, P. G45R on nonstructural protein 1 of influenza A virus contributes to virulence by increasing the expression of proinflammatory cytokines in mice. Arch. Virol. 2017, 162, 45–55. [Google Scholar] [CrossRef]
- Seo, S.H.; Hoffmann, E.; Webster, R.G. Lethal H5N1 influenza viruses escape host anti-viral cytokine responses. Nat. Med. 2002, 8, 950–954. [Google Scholar] [CrossRef]
- Lipatov, A.S.; Andreansky, S.; Webby, R.J.; Hulse, D.J.; Rehg, J.E.; Krauss, S.; Perez, D.R.; Doherty, P.C.; Webster, R.G.; Sangster, M.Y. Pathogenesis of Hong Kong H5N1 influenza virus NS gene reassortants in mice: The role of cytokines and B- and T-cell responses. J. Gen. Virol. 2005, 86, 1121–1130. [Google Scholar] [CrossRef]
- Ramos, I.; Fernandez-Sesma, A. Modulating the Innate Immune Response to Influenza A Virus: Potential Therapeutic Use of Anti-Inflammatory Drugs. Front. Immunol. 2015, 6, 361. [Google Scholar] [CrossRef]
- Nogales, A.; Martinez-Sobrido, L. Reverse genetics approaches for the development of influenza vaccines. Int. J. Mol. Sci. 2016, 18, 20. [Google Scholar] [CrossRef]
- Belongia, E.A.; Kieke, B.A.; Donahue, J.G.; Greenlee, R.T.; Balish, A.; Foust, A.; Lindstrom, S.; Shay, D.K. Effectiveness of inactivated influenza vaccines varied substantially with antigenic match from the 2004–2005 season to the 2006–2007 season. J. Infect Dis. 2009, 199, 159–167. [Google Scholar] [CrossRef]
- Osterholm, M.T.; Kelley, N.S.; Sommer, A.; Belongia, E.A. Efficacy and effectiveness of influenza vaccines: A systematic review and meta-analysis. Lancet Infect. Dis. 2012, 12, 36–44. [Google Scholar] [CrossRef]
- Kim, H.; Webster, R.G.; Webby, R.J. Influenza Virus: Dealing with a Drifting and Shifting Pathogen. Viral Immunol. 2018, 31, 174–183. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Anderson, C.S.; Yang, H.; Holden-Wiltse, J.; Fitzgerald, T.; Treanor, J.J.; Topham, D.J. Directed selection of influenza virus produces antigenic variants that match circulating human virus isolates and escape from vaccine-mediated immune protection. Immunology 2016, 148, 160–173. [Google Scholar] [CrossRef]
- Wong, S.S.; Webby, R.J. Traditional and new influenza vaccines. Clin. Microbiol. Rev. 2013, 26, 476–492. [Google Scholar] [CrossRef]
- Belshe, R.B.; Edwards, K.M.; Vesikari, T.; Black, S.V.; Walker, R.E.; Hultquist, M.; Kemble, G.; Connor, E.M.; Group, C.-T.C.E.S. Live attenuated versus inactivated influenza vaccine in infants and young children. N. Engl. J. Med. 2007, 356, 685–696. [Google Scholar] [CrossRef]
- Cox, M.M.; Patriarca, P.A.; Treanor, J. FluBlok, a recombinant hemagglutinin influenza vaccine. Influenza Other Respir Viruses 2008, 2, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.; Zhou, H.; Kemble, G.; Jin, H. The cold adapted and temperature sensitive influenza A/Ann Arbor/6/60 virus, the master donor virus for live attenuated influenza vaccines, has multiple defects in replication at the restrictive temperature. Virology 2008, 380, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Li, Y.; Belser, J.A.; Pearce, M.B.; Schmolke, M.; Subba, A.X.; Shi, Z.; Zaki, S.R.; Blau, D.M.; Garcia-Sastre, A.; et al. NS-based live attenuated H1N1 pandemic vaccines protect mice and ferrets. Vaccine 2010, 28, 8015–8025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossler, C.; Groiss, F.; Wolzt, M.; Wolschek, M.; Seipelt, J.; Muster, T. Phase I/II trial of a replication-deficient trivalent influenza virus vaccine lacking NS1. Vaccine 2013, 31, 6194–6200. [Google Scholar] [CrossRef]
- Du, Y.; Xin, L.; Shi, Y.; Zhang, T.H.; Wu, N.C.; Dai, L.; Gong, D.; Brar, G.; Shu, S.; Luo, J.; et al. Genome-wide identification of interferon-sensitive mutations enables influenza vaccine design. Science 2018, 359, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.A.; Wills, N.M.; Xiao, Y.L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An Overlapping Protein-Coding Region in Influenza A Virus Segment 3 Modulates the Host Response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resa-Infante, P.; Jorba, N.; Coloma, R.; Ortin, J. The influenza virus RNA synthesis machine: Advances in its structure and function. RNA Biol. 2011, 8, 207–215. [Google Scholar] [CrossRef]
- Firth, A.E.; Jagger, B.W.; Wise, H.M.; Nelson, C.C.; Parsawar, K.; Wills, N.M.; Napthine, S.; Taubenberger, J.K.; Digard, P.; Atkins, J.F. Ribosomal frameshifting used in influenza A virus expression occurs within the sequence UCC_UUU_CGU and is in the +1 direction. Open Biol. 2012, 2, 120109. [Google Scholar] [CrossRef]
- Shi, M.; Jagger, B.W.; Wise, H.M.; Digard, P.; Holmes, E.C.; Taubenberger, J.K. Evolutionary conservation of the PA-X open reading frame in segment 3 of influenza A virus. J. Virol. 2012, 86, 12411–12413. [Google Scholar] [CrossRef]
- Hayashi, T.; MacDonald, L.A.; Takimoto, T. Influenza A Virus Protein PA-X Contributes to Viral Growth and Suppression of the Host Antiviral and Immune Responses. J. Virol. 2015, 89, 6442–6452. [Google Scholar] [CrossRef] [Green Version]
- Khaperskyy, D.A.; McCormick, C. Timing Is Everything: Coordinated Control of Host Shutoff by Influenza A Virus NS1 and PA-X Proteins. J. Virol. 2015, 89, 6528–6531. [Google Scholar] [CrossRef] [Green Version]
- Khaperskyy, D.A.; Schmaling, S.; Larkins-Ford, J.; McCormick, C.; Gaglia, M.M. Selective Degradation of Host RNA Polymerase II Transcripts by Influenza A Virus PA-X Host Shutoff Protein. PLoS Pathog. 2016, 12, e1005427. [Google Scholar] [CrossRef]
- Hu, J.; Ma, C.; Liu, X. PA-X: A key regulator of influenza A virus pathogenicity and host immune responses. Med. Microbiol. Immunol. 2018, 207, 1–15. [Google Scholar] [CrossRef]
- Hu, J.; Mo, Y.Q.; Wang, X.Q.; Gu, M.; Hu, Z.L.; Zhong, L.; Wu, Q.W.; Hao, X.L.; Hu, S.L.; Liu, W.B.; et al. PA-X Decreases the Pathogenicity of Highly Pathogenic H5N1 Influenza A Virus in Avian Species by Inhibiting Virus Replication and Host Response. J. Virol. 2015, 89, 4126–4142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bavagnoli, L.; Cucuzza, S.; Campanini, G.; Rovida, F.; Paolucci, S.; Baldanti, F.; Maga, G. The novel influenza A virus protein PA-X and its naturally deleted variant show different enzymatic properties in comparison to the viral endonuclease PA. Nucleic Acids Res. 2015, 43, 9405–9417. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.J.; Xu, G.L.; Sun, Y.P.; Qi, L.; Wang, J.L.; Kong, W.L.; Sun, H.L.; Pu, J.; Chang, K.C.; Liu, J.H. PA-X is a virulence factor in avian H9N2 influenza virus. J. Gen. Virol. 2015, 96, 2587–2594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Yu, H.; Li, Y.; Ma, J.; Lang, Y.; Duff, M.; Henningson, J.; Liu, Q.; Li, Y.; Nagy, A.; et al. Impacts of different expressions of PA-X protein on 2009 pandemic H1N1 virus replication, pathogenicity and host immune responses. Virology 2017, 504, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Chaimayo, C.; McGuinness, J.; Takimoto, T. Critical Role of the PA-X C-Terminal Domain of Influenza A Virus in Its Subcellular Localization and Shutoff Activity. J. Virol. 2016, 90, 7131–7141. [Google Scholar] [CrossRef] [Green Version]
- Desmet, E.A.; Bussey, K.A.; Stone, R.; Takimoto, T. Identification of the N-terminal domain of the influenza virus PA responsible for the suppression of host protein synthesis. J. Virol. 2013, 87, 3108–3118. [Google Scholar] [CrossRef]
- Oishi, K.; Yamayoshi, S.; Kawaoka, Y. Mapping of a Region of the PA-X Protein of Influenza A Virus That Is Important for Its Shutoff Activity. J. Virol. 2015, 89, 8661–8665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Sun, H.; Hu, J.; Qi, L.; Wang, J.; Xiong, X.; Wang, Y.; He, Q.; Lin, Y.; Kong, W.; et al. Twenty amino acids at the C-terminus of PA-X are associated with increased influenza A virus replication and pathogenicity. J. Gen. Virol. 2015, 96, 2036–2049. [Google Scholar] [CrossRef] [Green Version]
- Oishi, K.; Yamayoshi, S.; Kawaoka, Y. Identification of novel amino acid residues of influenza virus PA-X that are important for PA-X shutoff activity by using yeast. Virology 2018, 516, 71–75. [Google Scholar] [CrossRef]
- Gao, H.; Sun, Y.; Hu, J.; Qi, L.; Wang, J.; Xiong, X.; Wang, Y.; He, Q.; Lin, Y.; Kong, W.; et al. The contribution of PA-X to the virulence of pandemic 2009 H1N1 and highly pathogenic H5N1 avian influenza viruses. Sci. Rep. 2015, 5, 8262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Mo, Y.; Gao, Z.; Wang, X.; Gu, M.; Liang, Y.; Cheng, X.; Hu, S.; Liu, W.; Liu, H.; et al. PA-X-associated early alleviation of the acute lung injury contributes to the attenuation of a highly pathogenic H5N1 avian influenza virus in mice. Med. Microbiol. Immunol. 2016, 205, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Rodriguez, L.; DeDiego, M.L.; Topham, D.J.; Martinez-Sobrido, L. Interplay of PA-X and NS1 Proteins in Replication and Pathogenesis of a Temperature-Sensitive 2009 Pandemic H1N1 Influenza A Virus. J. Virol. 2017, 91, JVI-00720. [Google Scholar] [CrossRef] [PubMed]
- Aydillo, T.; Ayllon, J.; Pavlisin, A.; Martinez-Romero, C.; Tripathi, S.; Mena, I.; Moreira-Soto, A.; Vicente-Santos, A.; Corrales-Aguilar, E.; Schwemmle, M.; et al. Specific Mutations in the PB2 Protein of Influenza A Virus Compensate for the Lack of Efficient Interferon Antagonism of the NS1 Protein of Bat Influenza A-Like Viruses. J. Virol. 2018, 92, JVI-02021. [Google Scholar] [CrossRef] [PubMed]
- Toback, S.L.; Levin, M.J.; Block, S.L.; Belshe, R.B.; Ambrose, C.S.; Falloon, J. Quadrivalent Ann Arbor strain live-attenuated influenza vaccine. Expert Rev. Vaccines 2012, 11, 1293–1303. [Google Scholar] [CrossRef] [Green Version]
- Caspard, H.; Mallory, R.M.; Yu, J.; Ambrose, C.S. Live-attenuated influenza vaccine effectiveness in children from 2009 to 2015–2016: A systematic review and meta-analysis. Open Forum Infect. Dis. 2017, 4, ofx111. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host Factor a | NS1 Domain b | Protein Function c | Reference |
|---|---|---|---|
| TRIM25 | RBD and ED | Ubiquitin E3 ligase and ISG15 E3 ligase. Modification of RIG-I by poly-ubiquitin chains synthesized by TRIM25 is essential for RIG-I activation | [91,92] |
| Riplet | RBD and ED | E2-dependent E3 ubiquitin-protein ligase. Modification of RIG-I by poly-ubiquitin chains synthesized by Riplet is essential for RIG-I activation | [92] |
| RIG-I | RBD | Cytoplasmic PRR of viral nucleic acids, activating innate immune responses | [96] |
| hPAF1C | CTT | Transcription of Hox and Wnt genes and histone modifications | [97] |
| IKKβ | CTT | Serine kinase involved in NF-κB activation and IFN and proinflammatory responses | [98] |
| IRF3 | Indirect effect | Transcriptional regulator factor of type I IFN-dependent immune responses | [99] |
| AP-1 | Indirect effect | Transcription factor involved in IFN and pro-inflammatory cytokines induction | [100] |
| A20 | Indirect effect | Ubiquitin-editing enzyme that contains both ubiquitin ligase and deubiquitinase activities. Suppresses IRF3-mediated IFN induction | [94,95] |
| PKR | RBD and ED | IFN-induced dsRNA-dependent serine/threonine-protein kinase leading to protein translation inhibition | [101,102,103] |
| RNase L | Indirect effect | Endoribonuclease leading to host gene expression inhibition by mRNA degradation | [87] |
| PACT | NS1 | Activation of EIF2AK2/PKR | [101,104] |
| OAS | RBD and indirect effect | Polymerizes higher oligomers of 2’-5’-oligoadenylates that bind to RNaseL leading to its activation | [51,87] |
| NLRP3 | ED and indirect effect | Sensor component of the NLRP3 inflammasome, leading to inflammatory responses | [105,106,107] |
| CPSF30 | RBD and ED | Processing of mRNAs, necessary for host gene protein expression | [62,72,74,108,109,110] |
| PABPII | CTT | Formation of mRNA precursors adding a poly(A) tail, necessary for gene expression | [111,112] |
| eIF4GI | RBD and ED | Recognition of the mRNA cap, ATP-dependent unwinding of 5’-terminal secondary structure and recruitment of mRNA to the ribosome. | [113] |
| p85β | ED | PI3K subunit | [114,115] |
| NXF1 | RBD and ED | Nuclear export of mRNA | [116] |
| RAE1 | RBD and ED | mRNA nucleocytoplasmic transport | [116] |
| P15 | ED | Nuclear export of mRNA | [116] |
| E1B-AP5 | RBD and ED | Transcriptional regulator | [116] |
| NS1 Amino Acid Residue | IAV | Reference |
|---|---|---|
| 184 | A/Udorn/72 H3N2 | [108] |
| 103 and 106 | A/Puerto Rico/8/34 H1N1 A/Hong Kong/483/97 H5N1 | [74,108,138] |
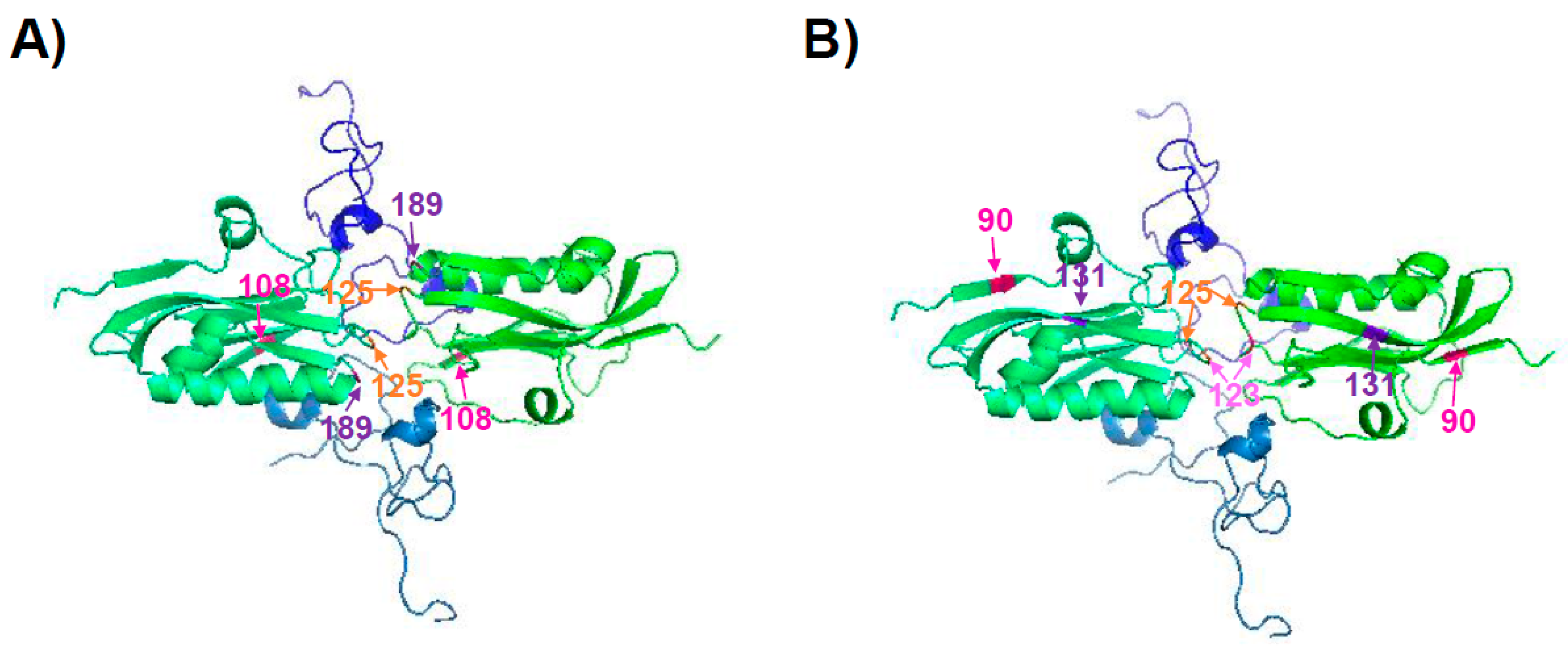
| 108, 125, 189, 55, 90, 123, 125, 131, and 205 | A/California/04/09 H1N1 | [125,126] |
| 106 | A/Shanghai/1/2013 H7N9 A/Shanghai/2/2013 H7N9 | [135] |
| 186 | A/canine/NY/dog23/2009 H3N8 A/equine/Ohio/1/2003 H3N8 | [62,72] |
| 64, 189, and 194 | A/Victoria/361/2011 H3N2 A/Perth/16 H3N2 | [83,84] |
| Properties | pH1N1 LAIV (ref. [174]) | pH1N1 WT (ref. [71]) | ||||||
|---|---|---|---|---|---|---|---|---|
| PAWT NS1WT | PAMUT NS1WT | PAWT NS1MUT | PAMUT NS1MUT | PAWT NS1WT | PAMUT NS1WT | PAWT NS1MUT | PAMUT NS1MUT | |
| Inhibition of host gene expression a | + − | − − | + + | − + | + − | − − | + + | − + |
| Pathogenicity in mice/ Viral replication in mice lungs | +++/ +++ | −/ − | −/ − | +++/ +++ | ++/ + | +++/ +++ | +/ + | +++/ +++ |
| Induction of innate immune response in mice b/Induction of humoral responses | +++/ +++ | −/ − | −/ − | +++/ +++ | +/ ND | ++/ ND | +/ ND | +++/ ND |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nogales, A.; Martinez-Sobrido, L.; Topham, D.J.; DeDiego, M.L. Modulation of Innate Immune Responses by the Influenza A NS1 and PA-X Proteins. Viruses 2018, 10, 708. https://doi.org/10.3390/v10120708
Nogales A, Martinez-Sobrido L, Topham DJ, DeDiego ML. Modulation of Innate Immune Responses by the Influenza A NS1 and PA-X Proteins. Viruses. 2018; 10(12):708. https://doi.org/10.3390/v10120708
Chicago/Turabian StyleNogales, Aitor, Luis Martinez-Sobrido, David J. Topham, and Marta L. DeDiego. 2018. "Modulation of Innate Immune Responses by the Influenza A NS1 and PA-X Proteins" Viruses 10, no. 12: 708. https://doi.org/10.3390/v10120708
APA StyleNogales, A., Martinez-Sobrido, L., Topham, D. J., & DeDiego, M. L. (2018). Modulation of Innate Immune Responses by the Influenza A NS1 and PA-X Proteins. Viruses, 10(12), 708. https://doi.org/10.3390/v10120708