Optimizing the Targeting of Mouse Parvovirus 1 to Murine Melanoma Selects for Recombinant Genomes and Novel Mutations in the Viral Capsid Gene

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses, Cells, and Selection of MPV1 on B16F10 Cells
2.2. Viral Sequencing
2.3. Viral Subcloning, Recovery, Purification, and Quantification
2.4. Analysis of Viral Infection Efficiency
2.5. Analysis of Viral Gene Expression and DNA Replication
2.6. Modeling of MPV1 Structures
2.7. GenBank Accession Numbers
3. Results
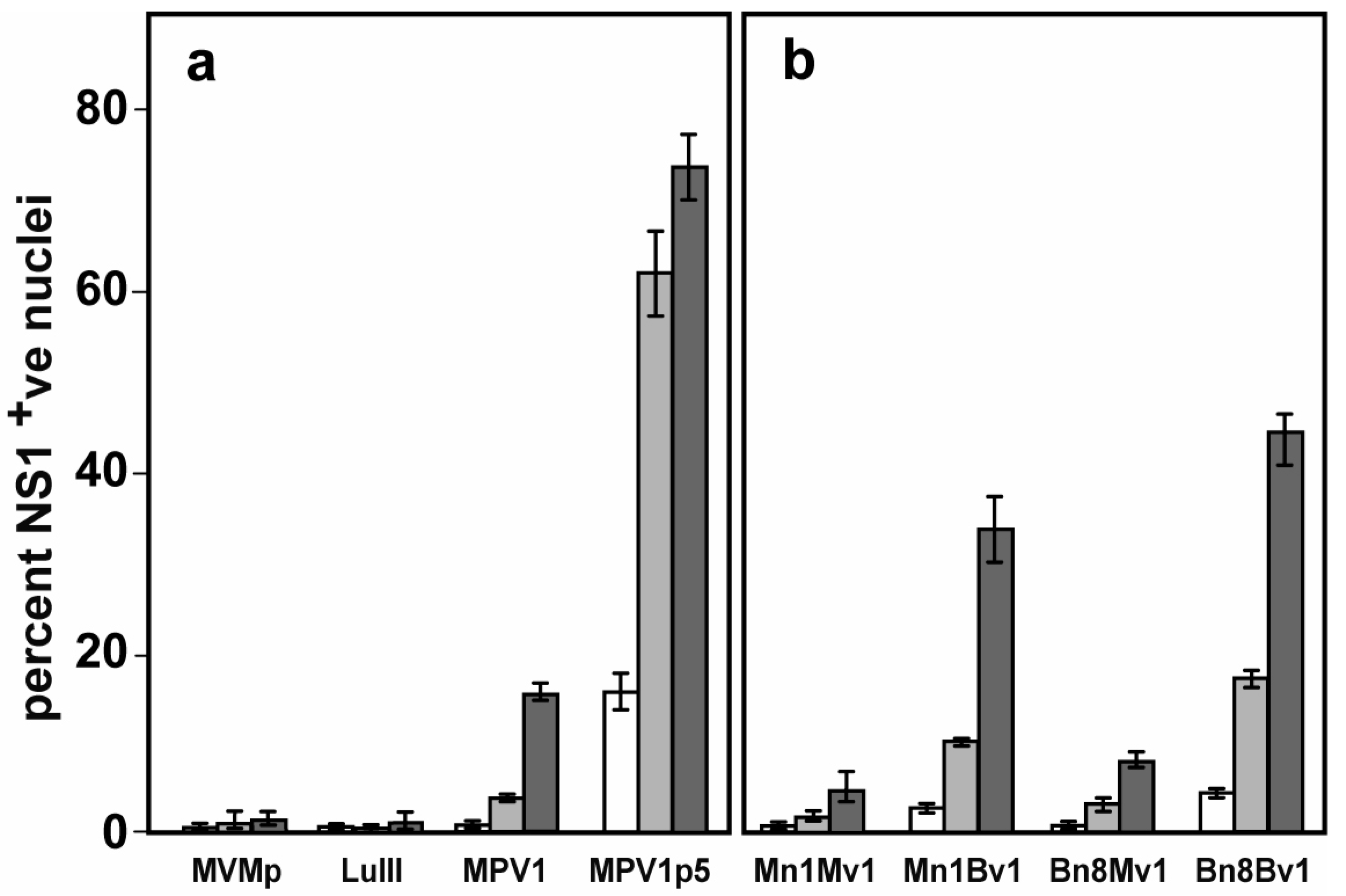
3.1. Screening for Viruses That Target Murine Melanoma
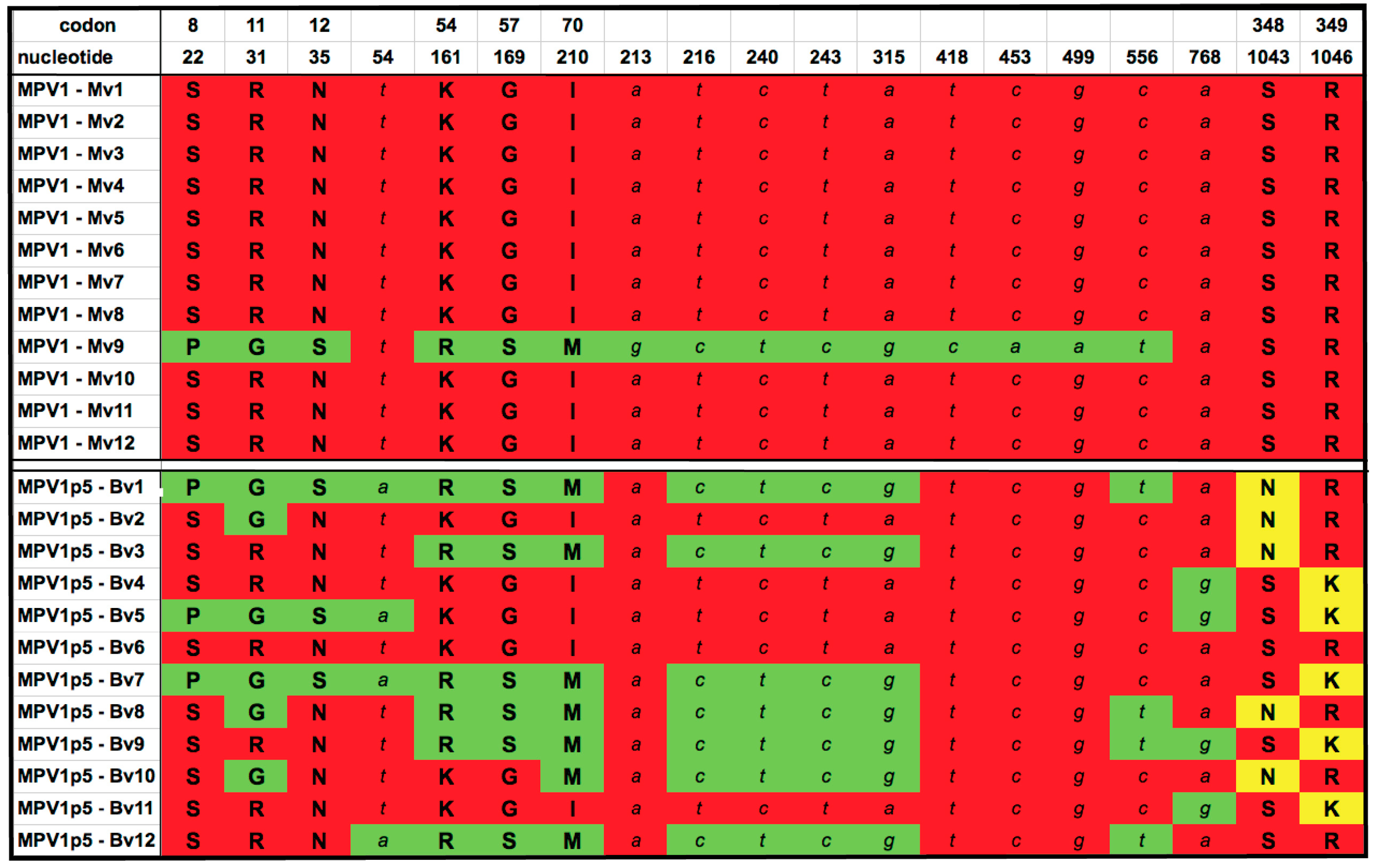
3.2. Genetic Analysis of the Parental and Selected Viruses
3.3. Mapping the Element(s) That Enhance Growth in B16F10 Melanoma Cells
3.4. Determining the Contribution of the 3′UTR and the Unique VP2 Mutations
3.5. Mapping the Enhancing Element(s) Using Chimeric Viruses
3.6. Biochemical Correlates of the Enhancement of Infectivity
4. Discussion
4.1. Targeted Evolution of the MPV1 Capsid
4.2. Localizing the Enhancing Changes within the Capsid Shell
Acknowledgments
Author Contributions
Conflicts of Interest
References
- ACS Melanoma Statistics. Available online: http://www.cancer.org/cancer/skincancer-melanoma/detailedguide/melanoma-skin-cancer-key-statistics (accessed on 27th January 2018).
- Klebanoff, C.A.; Acquavella, N.; Yu, Z.; Restifo, N.P. Therapeutic cancer vaccines: Are we there yet? Immunol. Rev. 2011, 239, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Sarnaik, A.A.; Weber, J.S. Recent advances using anti-CTLA-4 for the treatment of melanoma. Cancer J. 2009, 15, 169–173. [Google Scholar] [PubMed]
- Ibrahim, S.F.; Sambandan, D.; Ratner, D. Immunotherapy for Cutaneous Malignancy. Dermatol. Surg. 2011, 37, 1377–1393. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. Cell transfer immunotherapy for metastatic solid cancer-what clinicians need to know. Nat. Rev. Clin. Oncol. 2011, 8, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Antionia, S.J.; Vansteenkiste, J.F.; Moon, E. Immunotherapy: Beyond anti-PD-1 & anti-PD-L1 therapies. ASCO Educ. Book 2016, 35, e450–e458. [Google Scholar] [CrossRef]
- Herzberg, B.; Fisher, D.E. Metastatic melanoma and immunotherapy. Clin. Immunol. 2016, 172, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Merelli, B.; Massi, D.; Cattaneo, L.; Mandala, M. Targeting the PD1/PD-L1 axis in melanoma: Biological rationale, clinical challenges and opportunities. Crit. Rev. Oncol. Hematol. 2014, 89, 140–165. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Okuyama, R. Immunotherapy for advanced melanoma: Current knowledge and future directions. J. Dermatol. Sci. 2016, 83, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.A.; Peggs, K.S.; Simpson, T.R.; Allison, J.P. Shifting the equilibrium in cancer immunoediting: From tumor tolerance to eradication. Immunol. Rev. 2011, 241, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.E. Viruses with oncolytic properties and their adaptation to tumors. Ann. N. Y. Acad. Sci. 1952, 54, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef] [PubMed]
- Sivendran, S.; Pan, M.; Kaufman, H.L.; Saenger, Y. Herpes simplex virus oncolytic vaccine therapy in melanoma. Expert Opin. Biol. Ther. 2010, 10, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Kohlhapp, F.J.; Kaufman, H.L. Molecular Pathways: Mechanism of Action for Talimogene Laherparepvec, a New Oncolytic Virus Immunotherapy. Clin. Cancer Res. 2016, 22, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Wen, Z.; Li, C.; Lu, H.; Tian, M.; Jin, K.; Sun, L.; Gao, P.; Yang, E.; et al. Potent anti-tumor effects of a dual specific oncolytic adenovirus expressing apoptin in vitro and in vivo. Mol. Cancer 2010, 9, 10. [Google Scholar] [PubMed]
- Lazar, I.; Yaacov, B.; Shiloach, T.; Eliahoo, E.; Kadouri, L.; Lotem, M.; Perlman, R.; Zakay-Rones, Z.; Panet, A.; Ben-Yehuda, D. The oncolytic activity of Newcastle disease virus NDV-HUJ on chemoresistant primary melanoma cells is dependent on the proapoptotic activity of the inhibitor of apoptosis protein Livin. J. Virol. 2010, 84, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Vigil, A.; Kelly, K.; Garcia-Sastre, A.; Fong, Y. Genetically engineered Newcastle disease virus for malignant melanoma therapy. Gene Ther. 2009, 16, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Errington, F.; Ilett, E.J.; Morgan, R.S.; Scott, K.J.; Kottke, T.; Thompson, J.; Morrison, E.E.; Harrington, K.J.; Pandha, H.S.; et al. Tumor infection by oncolytic reovirus primes adaptive antitumor immunity. Clin. Cancer Res. 2008, 14, 7358–7366. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Wang, H.; Kottke, T.; Diaz, R.M.; Willmon, C.; Hudacek, A.; Thompson, J.; Parato, K.; Bell, J.; Naik, J.; et al. Loading of oncolytic vesicular stomatitis virus onto antigen-specific T cells enhances the efficacy of adoptive T-cell therapy of tumors. Gene Ther. 2008, 15, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.L.; Doty, R.; Tosic, V.; Liu, J.; Kranz, D.M.; McFadden, G.; Macneill, A.L.; Roy, E.J. Myxoma virus combined with rapamycin treatment enhances adoptive T cell therapy for murine melanoma brain tumors. Cancer Immunol. Immunother. 2011, 60, 1461. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Kiprianova, I.; Ayache, A.; Koch, R.; Herrero, Y.C.M.; Deleu, L.; Sommer, C.; Thomas, N.; Rommelaere, J.; Schlehofer, J.R. Regression of advanced rat and human gliomas by local or systemic treatment with oncolytic parvovirus H-1 in rat models. Neuro Oncol. 2010, 12, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Raykov, Z.; Grekova, S.; Galabov, A.S.; Balboni, G.; Koch, U.; Aprahamian, M.; Rommelaere, J. Combined oncolytic and vaccination activities of parvovirus H-1 in a metastatic tumor model. Oncol. Rep. 2007, 17, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family Parvoviridae. Arch. Virol. 2014, 159, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. Parvoviruses: Small Does Not Mean Simple. Annu. Rev. Virol. 2014, 1, 517–537. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. Parvovirus diversity and DNA damage responses. Cold Spring Harbor Perspect. Biol. 2013, 5, a012989. [Google Scholar] [CrossRef] [PubMed]
- Vollmers, E.M.; Tattersall, P. Distinct host cell fates for human malignant melanoma targeted by oncolytic rodent parvoviruses. Virology 2013, 446, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Humrich, J.Y.; Thuman, P.; Sauter, B.; Schuler, G.; Jenne, L. The highly attenuated vaccinia virus strain modified virus Ankara induces apoptosis in melanoma cells and allows bystander dendritic cells to generate a potent anti-tumoral immunity. Clin. Exp. Immunol. 2006, 146, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Grekova, S.; Aprahamian, M.; Giese, N.; Schmitt, S.; Giese, T.; Falk, C.S.; Daeffler, L.; Cziepluch, C.; Rommelaere, J.; Raykov, Z. Immune cells participate in the oncosuppressive activity of parvovirus H-1PV and are activated as a result of their abortive infection with this agent. Cancer Biol. Ther. 2011, 10, 1280–1289. [Google Scholar] [CrossRef]
- Prestwich, R.J.; Errington, F.; Diaz, R.M.; Pandha, H.S.; Harrington, K.J.; Melcher, A.A.; Vile, R.G. The case of oncolytic viruses versus the immune system: Waiting on the judgment of Solomon. Hum. Gene Ther. 2009, 20, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Houben, R.; Schrama, D.; Voigt, H.; Ugurel, S.; Reisfeld, R.A. Mouse models for melanoma: A personal perspective. Exp. Dermatol. 2010, 19, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cotmore, S.F.; Tattersall, P. Parvoviral left-end hairpin ears are essential during infection for establishing a functional intranuclear transcription template and for efficient progeny genome encapsidation. J. Virol. 2013, 87, 10501–10514. [Google Scholar] [CrossRef] [PubMed]
- Ball-Goodrich, L.J.; Johnson, E. Molecular characterization of a newly recognized mouse parvovirus. J. Virol. 1994, 68, 6476–6486. [Google Scholar] [PubMed]
- Tattersall, P.; Bratton, J. Reciprocal productive and restrictive virus-cell interactions of immunosuppressive and prototype strains of minute virus of mice. J. Virol. 1983, 46, 944–955. [Google Scholar] [PubMed]
- Li, L.; Cotmore, S.F.; Tattersall, P. Maintenance of the flip sequence orientation of the ears in the parvoviral left-end hairpin is a nonessential consequence of the critical asymmetry in the hairpin stem. J. Virol. 2012, 86, 12187–12197. [Google Scholar] [CrossRef] [PubMed]
- Reed, S.E.; Staley, E.M.; Mayginnes, J.P.; Pintel, D.J.; Tullis, G.E. Transfection of mammalian cells using linear polyethylenimine is a simple and effective means of producing recombinant adeno-associated virus vectors. J. Virol. Methods 2006, 138, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Li, J.; Samulski, R.J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998, 72, 2224–2232. [Google Scholar] [PubMed]
- Cotmore, S.F.; Hafenstein, S.; Tattersall, P. Depletion of virion-associated divalent cations induces parvovirus minute virus of mice to eject its genome in a 3′-to-5′ direction from an otherwise intact viral particle. J. Virol. 2010, 84, 1945–1956. [Google Scholar] [CrossRef] [PubMed]
- Farr, G.A.; Tattersall, P. A conserved leucine that constricts the pore through the capsid fivefold cylinder plays a central role in parvoviral infection. Virology 2004, 323, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Yeung, D.E.; Brown, G.W.; Tam, P.; Russnak, R.H.; Wilson, G.; Clark-Lewis, I.; Astell, C.R. Monoclonal antibodies to the major nonstructural nuclear protein of minute virus of mice. Virology 1991, 181, 35–45. [Google Scholar] [CrossRef]
- Ruiz, Z.; Mihaylov, I.S.; Cotmore, S.F.; Tattersall, P. Recruitment of DNA replication and damage response proteins to viral replication centers during infection with NS2 mutants of Minute Virus of Mice (MVM). Virology 2011, 410, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. The NS-1 polypeptide of the autonomous parvovirus MVM is a nuclear phosphoprotein. Virus Res. 1986, 4, 243–250. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Tattersall, P. Alternate splicing in a parvoviral nonstructural gene links a common amino-terminal sequence to downstream domains which confer radically different localization and turnover characteristics. Virology 1990, 177, 477–487. [Google Scholar] [CrossRef]
- D’Abramo, A.M.; Ali, A.A.; Wang, F.; Cotmore, S.F.; Tattersall, P. Host range mutants of Minute Virus of Mice with a single VP2 amino acid change require additional silent mutations that regulate NS2 accumulation. Virology 2005, 340, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Kontou, M.; Govindaswamy, L.; Nam, H.-J.; Bryant, N.; Llamas-Saiz, A.L.; Foces-Foces, C.; Hernando, E.; Rubio, M.-P.; McKenna, R.; Almendral, J.M.; et al. Structural determinants of tissue tropism and in vivo pathogenicity for the parvovirus minute virus of mice. J. Virol. 2005, 79, 10931–10943. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Tripp, M.; Shepherd, C.M.; Borelli, I.A.; Venkataraman, S.; Lander, G.; Natarajan, P.; Johnson, J.E.; Brooks, C.L., 3rd; Reddy, V.S. VIPERdb2: An enhanced and web API enabled relational database for structural virology. Nucleic Acids Res. 2009, 37, D436–D442. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Rossmann, M.G. Interpretation of electron density with stereographic roadmap projections. J. Struct. Biol. 2007, 158, 182–187. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific LLC: San Carlos, CA, USA, 2002. [Google Scholar]
- Mattei, L.M.; Cotmore, S.F.; Tattersall, P.; Iwasaki, A. Parvovirus evades interferon-dependent viral control in primary mouse embryonic fibroblasts. Virology 2013, 442, 20–27. [Google Scholar] [CrossRef] [PubMed]
- López-Bueno, A.; Mateu, M.G.; Almendral, J.M. High mutant frequency in populations of a DNA virus allows evasion from antibody therapy in an immunodeficient host. J. Virol. 2003, 77, 2701–2708. [Google Scholar] [CrossRef] [PubMed]
- López-Bueno, A.; Segovia, J.C.; Bueren, J.A.; O’Sullivan, M.G.; Wang, F.; Tattersall, P.; Almendral, J.M. Evolution to pathogenicity of the parvovirus minute virus of mice in immunodeficient mice involves genetic heterogeneity at the capsid domain that determines tropism. J. Virol. 2008, 82, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. Encapsidation of minute virus of mice DNA: Aspects of the translocation mechanism revealed by the structure of partially packaged genomes. Virology 2005, 336, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Paglino, J.; Tattersall, P. The parvoviral capsid controls an intracellular phase of infection essential for efficient killing of stepwise-transformed human fibroblasts. Virology 2011, 416, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Joh, J.; Proctor, M.L.; Ditslear, J.L.; King, W.W.; Sundberg, J.P.; Jenson, A.B.; Ghim, S.J. Epidemiological and phylogenetic analysis of institutional mouse parvoviruses. Exp. Mol. Pathol. 2013, 95, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Besselsen, D.G.; Romero, M.J.; Wagner, A.M.; Henderson, K.S.; Livingston, R.S. Identification of novel murine parvovirus strains by epidemiological analysis of naturally infected mice. J. Gen. Virol. 2006, 87 Pt 6, 1543–1556. [Google Scholar] [CrossRef] [PubMed]
- Agbandje-McKenna, M.; Llamas-Saiz, A.L.; Wang, F.; Tattersall, P.; Rossmann, M.G. Functional implications of the structure of the murine parvovirus, minute virus of mice. Structure 1998, 6, 1369–1381. [Google Scholar] [CrossRef]
- Maroto, B.; Valle, N.; Saffrich, R.; Almendral, J.M. Nuclear export of the nonenveloped parvovirus virion is directed by an unordered protein signal exposed on the capsid surface. J. Virol. 2004, 78, 10685–10694. [Google Scholar] [CrossRef] [PubMed]
- Engelsma, D.; Valle, N.; Fish, A.; Salome, N.; Almendral, J.M.; Fornerod, M. A supraphysiological nuclear export signal is required for parvovirus nuclear export. Mol. Biol. Cell 2008, 19, 2544–2552. [Google Scholar] [CrossRef] [PubMed]
- López-Bueno, A.; Valle, N.; Gallego, J.M.; Pérez, J.; Almendral, J.M. Enhanced cytoplasmic sequestration of the nuclear export receptor CRM1 by NS2 mutations developed in the host regulates parvovirus fitness. J. Virol. 2004, 78, 10674–10684. [Google Scholar] [CrossRef] [PubMed]
- Maroto, B.; Ramírez, J.C.; Almendral, J.M. Phosphorylation status of the parvovirus minute virus of mice particle: Mapping and biological relevance of the major phosphorylation sites. J. Virol. 2000, 74, 10892–10902. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; D’Abramo, A.M.; Ticknor, C.M.; Tattersall, P. Controlled conformational transitions in the MVM virion expose the VP1 N-terminus and viral genome without particle disassembly. Virology 1999, 254, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. Mutations at the Base of the Icosahedral Five-Fold Cylinders of Minute Virus of Mice Induce 3′-to-5′ Genome Uncoating and Critically Impair Entry Functions. J. Virol. 2012, 86, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Farr, G.A.; Zhang, L.G.; Tattersall, P. Parvoviral virions deploy a capsid-tethered lipolytic enzyme to breach the endosomal membrane during cell entry. Proc. Natl. Acad. Sci. USA 2005, 102, 17148–17153. [Google Scholar] [CrossRef] [PubMed]
- Zádori, Z.; Szelei, J.; Lacoste, M.C.; Li, Y.; Gariépy, S.; Raymond, P.; Allaire, M.; Nabi, I.R.; Tijssen, P. A viral phospholipase A2 is required for parvovirus infectivity. Dev. Cell 2001, 1, 291–302. [Google Scholar] [CrossRef]
- Aydemir, F.; Salganik, M.; Resztak, J.; Singh, J.; Bennett, A.; Agbandje-McKenna, M.; Muzyczka, N. Mutants at the 2-Fold Interface of Adeno-associated Virus Type 2 (AAV2) Structural Proteins Suggest a Role in Viral Transcription for AAV Capsids. J. Virol. 2016, 90, 7196–7204. [Google Scholar] [CrossRef] [PubMed]
- Govindasamy, L.; Gurda, B.L.; Halder, S.; van Vliet, K.; McKenna, R.; Cotmore, S.F.; Tattersall, P.; Agbandje-McKenna, M. MVM capsid dynamics associated with DNA packaging and VP2 externalization for maturation cleavage. In Proceedings of the XIII International Parvovirus Workshop, Helsinki, Finland, 20–24 June 2010. [Google Scholar]
- Halder, S.; Cotmore, S.; Heimburg-Molinaro, J.; Smith, D.F.; Cummings, R.D.; Chen, X.; Trollope, A.J.; North, S.J.; Haslam, S.M.; Dell, A.; et al. Profiling of glycan receptors for minute virus of mice in permissive cell lines towards understanding the mechanism of cell recognition. PLoS ONE 2014, 9, e86909. [Google Scholar] [CrossRef] [PubMed]
- López-Bueno, A.; Rubio, M.-P.; Bryant, N.; McKenna, R.; Agbandje-McKenna, M.; Almendral, J.M. Host-selected amino acid changes at the sialic acid binding pocket of the parvovirus capsid modulate cell binding affinity and determine virulence. J. Virol. 2006, 80, 1563–1573. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marr, M.; D’Abramo, A.; Pittman, N.; Agbandje-McKenna, M.; Cotmore, S.; Tattersall, P. Optimizing the Targeting of Mouse Parvovirus 1 to Murine Melanoma Selects for Recombinant Genomes and Novel Mutations in the Viral Capsid Gene. Viruses 2018, 10, 54. https://doi.org/10.3390/v10020054
Marr M, D’Abramo A, Pittman N, Agbandje-McKenna M, Cotmore S, Tattersall P. Optimizing the Targeting of Mouse Parvovirus 1 to Murine Melanoma Selects for Recombinant Genomes and Novel Mutations in the Viral Capsid Gene. Viruses. 2018; 10(2):54. https://doi.org/10.3390/v10020054
Chicago/Turabian StyleMarr, Matthew, Anthony D’Abramo, Nikea Pittman, Mavis Agbandje-McKenna, Susan Cotmore, and Peter Tattersall. 2018. "Optimizing the Targeting of Mouse Parvovirus 1 to Murine Melanoma Selects for Recombinant Genomes and Novel Mutations in the Viral Capsid Gene" Viruses 10, no. 2: 54. https://doi.org/10.3390/v10020054
APA StyleMarr, M., D’Abramo, A., Pittman, N., Agbandje-McKenna, M., Cotmore, S., & Tattersall, P. (2018). Optimizing the Targeting of Mouse Parvovirus 1 to Murine Melanoma Selects for Recombinant Genomes and Novel Mutations in the Viral Capsid Gene. Viruses, 10(2), 54. https://doi.org/10.3390/v10020054