Biology of Porcine Parvovirus (Ungulate parvovirus 1)

{kind=link}
{kind=link}
Abstract
:1. Introduction
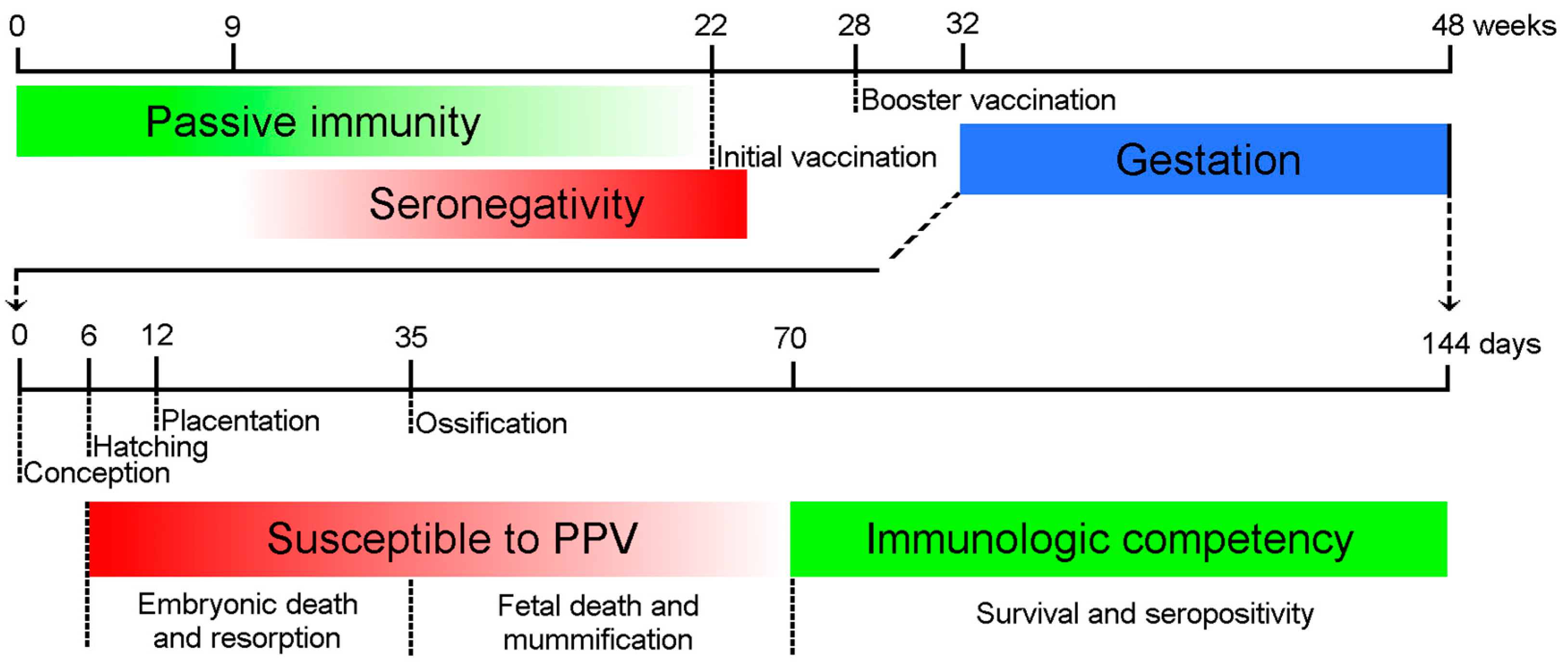
2. Pathogenesis
3. Virus-Cell Interaction
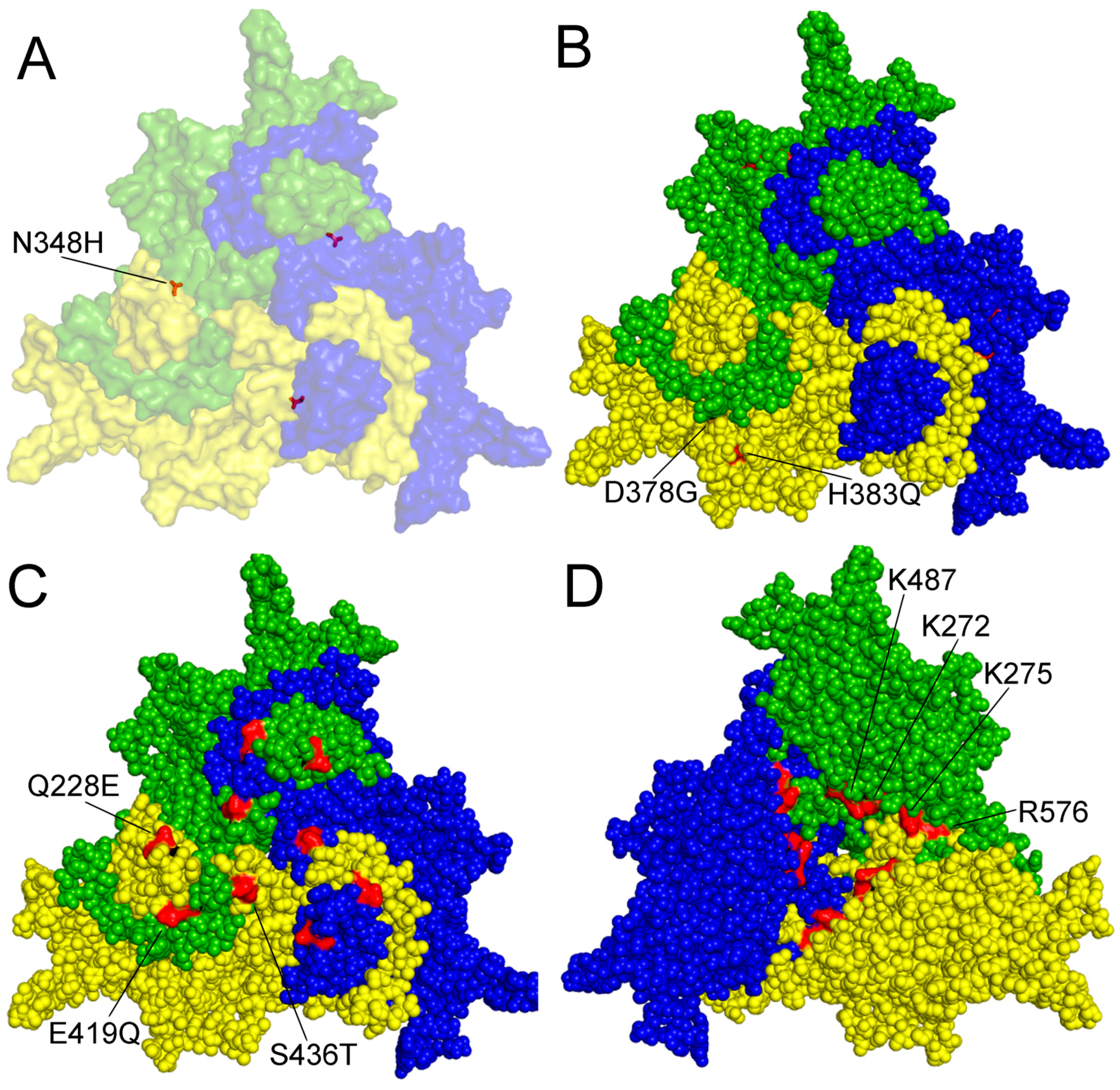
4. Genetic Variation and Evolution
5. Immunity and Prevention
6. Co-Infection with Circovirus
7. Detection and Isolation of PPV
Acknowledgments
Conflicts of Interest
References
- Cartwrigh, S.; Huck, R. Viruses isolated in association with herd infertility abortions and stillbirths in pigs. Vet. Rec. 1967, 81, 196–197. [Google Scholar]
- Cartwright, S.F.; Lucas, M.; Huck, R. A small haemagglutinating porcine DNA virus: I. Isolation and properties. J. Comp. Pathol. 1969, 79. [Google Scholar] [CrossRef]
- Johnson, R.; Collings, D. Experimental infection of piglets and pregnant gilts with a parvovirus. Vet. Rec. 1969, 85, 446–447. [Google Scholar] [CrossRef] [PubMed]
- Dunne, H.; Gobble, J.; Hokanson, J.; Kradel, D.; Bubash, G. Porcine reproductive failure associated with a newly identified “SMEDI” group of picorna viruses. Am. J. Vet. Res. 1965, 26, 1284–1297. [Google Scholar] [PubMed]
- Zeeuw, E.J.L.; Leinecker, N.; Herwig, V.; Selbitz, H.J.; Truyen, U. Study of the virulence and cross-neutralization capability of recent porcine parvovirus field isolates and vaccine viruses in experimentally infected pregnant gilts. J. Gen. Virol. 2007, 88, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Mengeling, W.; Cutlip, R. Pathogenesis of in utero infection: Experimental infection of five-week-old porcine fetuses with porcine parvovirus. Am. J. Vet. Res. 1975, 36, 1173–1177. [Google Scholar] [PubMed]
- Joo, H.; Donaldson-Wood, C.; Johnson, R. Observations on the pathogenesis of porcine parvovirus infection. Arch. Virol. 1976, 51, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Mengeling, W.L.; Paul, P.S.; Brown, T.T. Transplacental infection and embryonic death following maternal exposure to porcine parvovirus near the time of conception. Arch. Virol. 1980, 65, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Donaldson-Wood, C.; Joo, H.; Johnson, R. The effect on reproductive performance of porcine parvovirus infection in a susceptible pig herd. Vet. Rec. 1977, 100, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Mengeling, W. Prevalence of porcine parvovirus-induced reproductive failure: An abattoir study. J. Am. Vet. Med. Assoc. 1978, 172, 1291–1294. [Google Scholar] [PubMed]
- Mengeling, W.L.; Lager, K.M.; Zimmerman, J.K.; Samarikermani, N.; Beran, G.W. A current assessment of the role of porcine parvovirus as a cause of fetal porcine death. J. Vet. Diagn. Investig. 1991, 3, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Donaldson-Wood, C.; JOD, H.; Allender, U. Observations on the epidemiology of porcine parvovirus. Aust. Vet. J. 1976, 52, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Kresse, J.I.; Taylor, W.D.; Stewart, W.W.; Eernisse, K.A. Parvovirus infection in pigs with necrotic and vesicle-like lesions. Vet. Microbiol. 1985, 10, 525–531. [Google Scholar] [CrossRef]
- Mengeling, W.L.; Pejsak, Z.; Paul, P.S. Biological assay of attenuated strain NADL-2 and virulent strain NADL-8 of porcine parvovirus. Am. J. Vet. Res. 1984, 45, 2403–2407. [Google Scholar] [PubMed]
- Wilhelm, S.; Zeeuw, E.J.L.; Selbitz, H.J.; Truyen, U. Tissue distribution of two field isolates and two vaccine strains of porcine parvovirus in foetal organs after experimental infection of pregnant sows as determined by real-time PCR. J. Vet. Med. B 2005, 52, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Mengeling, W.; Lager, K.; Vorwald, A. The effect of porcine parvovirus and porcine reproductive and respiratory syndrome virus on porcine reproductive performance. Anim. Reprod. Sci. 2000, 60, 199–210. [Google Scholar] [CrossRef]
- Oraveerakul, K.; Choi, C.S.; Molitor, T.W. Tissue tropisms of porcine parvovirus in swine. Arch. Virol. 1993, 130, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.S.; Mengeling, W.L.; Brown, T.T. Replication of Porcine Parvovirus in Peripheral-Blood Lymphocytes, Monocytes, and Peritoneal-Macrophages. Infect. Immun. 1979, 25, 1003–1007. [Google Scholar] [PubMed]
- Harding, M.J.; Molitor, T.W. Porcine parvovirus: Replication in and inhibition of selected cellular functions of swine alveolar macrophages and peripheral blood lymphocytes. Arch. Virol. 1988, 101, 105–117. [Google Scholar] [CrossRef] [PubMed]
- McKillen, J.; Hjertner, B.; Millar, A.; McNeilly, F.; Beldak, S.; Adair, B.; Allan, G. Molecular beacon real-time PCR detection of swine viruses. J. Virol. Methods 2007, 140, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.F.; Zhang, C.F.; Chen, C.M.; Cui, S.J. Real-time PCR to detect and analyze virulent PPV loads in artificially challenged sows and their fetuses. Vet. Microbiol. 2009, 138, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, M.; Fernandes, S.; Tijssen, P. Multiple pathways involved in porcine parvovirus cellular entry and trafficking toward the nucleus. J. Virol. 2010, 84, 7782–7792. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; D’Abramo, A.M.; Ticknor, C.M.; Tattersall, P. Controlled conformational transitions in the MVM virion expose the VP1 N-terminus and viral genome without particle disassembly. Virology 1999, 254, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Canaan, S.; Zadori, Z.; Ghomashchi, F.; Bollinger, J.; Sadilek, M.; Moreau, M.E.; Tijssen, P.; Gelb, M.H. Interfacial enzymology of parvovirus phospholipases A2. J. Biol. Chem. 2004, 279, 14502–14508. [Google Scholar] [CrossRef] [PubMed]
- Zádori, Z.; Szelei, J.; Lacoste, M.-C.; Li, Y.; Gariépy, S.; Raymond, P.; Allaire, M.; Nabi, I.R.; Tijssen, P. A viral phospholipase A 2 is required for parvovirus infectivity. Dev. Cell 2001, 1, 291–302. [Google Scholar] [CrossRef]
- Shivanna, V.; Kim, Y.; Chang, K.O. Ceramide formation mediated by acid sphingomyelinase facilitates endosomal escape of caliciviruses. Virology 2015, 483, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, M.; Bouchard-Levesque, V.; Fernandes, S.; Tijssen, P. Classic nuclear localization signals and a novel nuclear localization motif are required for nuclear transport of porcine parvovirus capsid proteins. J. Virol. 2014, 88, 11748–11759. [Google Scholar] [CrossRef] [PubMed]
- Riolobos, L.; Reguera, J.; Mateu, M.G.; Almendral, J.M. Nuclear transport of trimeric assembly intermediates exerts a morphogenetic control on the icosahedral parvovirus capsid. J. Mol. Biol. 2006, 357, 1026–1038. [Google Scholar] [CrossRef] [PubMed]
- Bar, S.; Daeffler, L.; Rommelaere, J.; Nuesch, J.P. Vesicular egress of non-enveloped lytic parvoviruses depends on gelsolin functioning. PLoS Pathog. 2008, 4, e1000126. [Google Scholar] [CrossRef] [PubMed]
- Pirtle, E.C. Titration of two porcine respiratory viruses in mammalian cell cultures by direct fluorescent antibody staining. Am. J. Vet. Res. 1974, 35, 249–250. [Google Scholar] [PubMed]
- Bergeron, J.; Hebert, B.; Tijssen, P. Genome organization of the Kresse strain of porcine parvovirus: Identification of the allotropic determinant and comparison with those of NADL-2 and field isolates. J. Virol. 1996, 70, 2508–2515. [Google Scholar] [PubMed]
- Fernandes, S.; Boisvert, M.; Tijssen, P. Genetic elements in the VP region of porcine parvovirus are critical to replication efficiency in cell culture. J. Virol. 2011, 85, 3025–3029. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, S.; Boisvert, M.; Szelei, J.; Tijssen, P. Differential replication of two porcine parvovirus strains in bovine cell lines ensues from initial DNA processing and NS1 expression. J. Gen. Virol. 2014, 95, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Huang, Y.; Du, Q.; Luo, X.M.; Zhang, L.; Zhao, X.M.; Tong, D.W. Porcine parvovirus infection induces apoptosis in PK-15 cells through activation of p53 and mitochondria-mediated pathway. Biochem. Biophys. Res. Commun. 2015, 456, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.M.; Xiang, H.L.; Bai, X.Y.; Fei, N.J.; Huang, Y.; Song, X.J.; Zhang, H.L.; Zhang, L.; Tong, D.W. Porcine parvovirus infection activates mitochondria-mediated apoptotic signaling pathway by inducing ROS accumulation. Virol. J. 2016, 13. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, I.; Toth, R.; Olasz, F.; Tijssen, P.; Zadori, Z. The SAT Protein of Porcine Parvovirus Accelerates Viral Spreading through Induction of Irreversible Endoplasmic Reticulum Stress. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Qiu, J. Parvovirus infection-induced cell death and cell cycle arrest. Future Virol. 2010, 5, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Zadori, Z.; Szelei, J.; Tijssen, P. SAT: A late NS protein of porcine parvovirus. J. Virol. 2005, 79, 13129–13138. [Google Scholar] [CrossRef] [PubMed]
- Lukashov, V.V.; Goudsmit, J. Evolutionary relationships among parvoviruses: Virus-host coevolution among autonomous primate parvoviruses and links between adeno-associated and avian parvoviruses. J. Virol. 2001, 75, 2729–2740. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bueno, A.; Villarreal, L.P.; Almendral, J.M. Parvovirus variation for disease: A difference with RNA viruses? Curr. Top. Microbiol. Immunol. 2006, 299, 349–370. [Google Scholar] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Streck, A.F.; Canal, C.W.; Truyen, U. Molecular epidemiology and evolution of porcine parvoviruses. Infect. Genet. Evol. 2015, 36, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Cadar, D.; Dan, A.; Tombacz, K.; Lorincz, M.; Kiss, T.; Becskei, Z.; Spinu, M.; Tuboly, T.; Csagola, A. Phylogeny and evolutionary genetics of porcine parvovirus in wild boars. Infect. Genet. Evol. 2012, 12, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Jóźwik, A.; Manteufel, J.; Selbitz, H.-J.; Truyen, U. Vaccination against porcine parvovirus protects against disease, but does not prevent infection and virus shedding after challenge infection with a heterologous virus strain. J. Gen. Virol. 2009, 90, 2437–2441. [Google Scholar] [CrossRef] [PubMed]
- Streck, A.F.; Homeier, T.; Foerster, T.; Truyen, U. Population dynamics and in vitro antibody pressure of porcine parvovirus indicate a decrease in variability. J. Gen. Virol. 2013, 94, 2050–2055. [Google Scholar] [CrossRef] [PubMed]
- Streck, A.F.; Bonatto, S.L.; Homeier, T.; Souza, C.K.; Goncalves, K.R.; Gava, D.; Canal, C.W.; Truyen, U. High rate of viral evolution in the capsid protein of porcine parvovirus. J. Gen. Virol. 2011, 92, 2628–2636. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Tao, Y.; Cui, J.; Suo, S.; Cong, Y.; Tijssen, P. Phylogeny and evolution of porcine parvovirus. Virus Res. 2013, 178, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Shangjin, C.; Cortey, M.; Segales, J. Phylogeny and evolution of the NS1 and VP1/VP2 gene sequences from porcine parvovirus. Virus Res. 2009, 140, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Martins Soares, R.; Cortez, A.; Heinemann, M.B.; Sakamoto, S.M.; Martins, V.G.; Bacci, M., Jr.; De Campos Fernandes, F.M.; Richtzenhain, L.J. Genetic variability of porcine parvovirus isolates revealed by analysis of partial sequences of the structural coding gene VP2. J. Gen. Virol. 2003, 84, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, P.; Ritzmann, M.; Selbitz, H.J.; Heinritzi, K.; Truyen, U. VP1 sequences of German porcine parvovirus isolates define two genetic lineages. J. Gen. Virol. 2006, 87, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, S.; Langeveld, J.; Botner, A.; Nielsen, J.; Schaaper, W.M.M.; Boshuizen, R.S.; Casal, J.I.; Hojrup, P.; Vela, C.; Meloen, R.; et al. Mapping the antigenic structure of porcine parvovirus at the level of peptides. Virus Res. 1998, 53, 163–173. [Google Scholar] [CrossRef]
- Hoelzer, K.; Shackelton, L.A.; Parrish, C.R. Presence and role of cytosine methylation in DNA viruses of animals. Nucleic Acids Res. 2008, 36, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Shackelton, L.A.; Parrish, C.R.; Holmes, E.C. Evolutionary basis of codon usage and nucleotide composition bias in vertebrate DNA viruses. J. Mol. Evol. 2006, 62, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Toth, R.; Meszaros, I.; Stefancsik, R.; Bartha, D.; Balint, A.; Zadori, Z. CpG Distribution and Methylation Pattern in Porcine Parvovirus. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Ladekjaer-Mikkelsen, A.S.; Nielsen, J. A longitudinal study of cell-mediated immunity in pigs infected with porcine parvovirus. Viral Immunol. 2002, 15, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.S.; Mengeling, W.L.; Brown, T.T. Effect of Vaccinal and Passive-Immunity on Experimental-Infection of Pigs with Porcine Parvovirus. Am. J. Vet. Res. 1980, 41, 1368–1371. [Google Scholar] [PubMed]
- Etoh, M.; Morishita, E.; Ochiai, M.; Watanabe, Y. Transitional antibodies and spontaneous infection in the porcine (Sus scrofa) parvo viral infections. Jpn. J. Swine Husb. Res. (Japan) 1979, 16, 237–239. [Google Scholar]
- Gava, D.; Souza, C.K.; Mores, T.J.; Argenti, L.E.; Streck, A.F.; Canal, C.W.; Bortolozzo, F.P.; Wentz, I. Dynamics of vanishing of maternally derived antibodies of Ungulate protoparvovirus 1 suggests an optimal age for gilts vaccination. Trop. Anim. Health Prod. 2017, 49, 1085–1088. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Fujisaki, Y. Immunizing effects of inactivated porcine parvovirus vaccine on piglets. Bull. Natl. Inst. Anim. Health 1976, 16, 81. [Google Scholar]
- Opriessnig, T.; Gerber, P.F.; Matzinger, S.R.; Meng, X.J.; Halbur, P.G. Markedly different immune responses and virus kinetics in littermates infected with porcine circovirus type 2 or porcine parvovirus type 1. Vet. Immunol. Immunopathol. 2017, 191, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Foerster, T.; Streck, A.F.; Speck, S.; Selbitz, H.-J.; Lindner, T.; Truyen, U. An inactivated whole-virus porcine parvovirus vaccine protects pigs against disease but does not prevent virus shedding even after homologous virus challenge. J. Gen. Virol. 2016, 97, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- Steiner, E.; Balmelli, C.; Gerber, H.; Summerfield, A.; McCullough, K. Cellular adaptive immune response against porcine circovirus type 2 in subclinically infected pigs. BMC Vet. Res. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Collings, D. Transplacental infection of piglets with a porcine parvovirus. Res. Vet. Sci. 1971, 12, 570–572. [Google Scholar] [PubMed]
- Cartwright, S.F.; Lucas, M.; Huck, R. A small haemagglutinating porcine DNA virus: II. Biological and serological studies. J. Comp. Pathol. 1971, 81, 145–155. [Google Scholar] [CrossRef]
- Li, X.; Zhu, L.; Liu, X.; Sun, X.; Zhou, Y.; Lang, Q.; Li, P.; Cai, Y.; Qiao, X.; Xu, Z. Differential expression of micrornas in porcine parvovirus infected porcine cell line. Virol. J. 2015, 12, 128. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.G.; Cui, L.C.; Tian, C.Y.; Zhang, G.C.; Huo, G.C.; Tang, L.J.; Li, Y.J. Immunogenicity of Recombinant Classic Swine Fever Virus CD8(+) T Lymphocyte Epitope and Porcine Parvovirus VP2 Antigen Coexpressed by Lactobacillus casei in Swine via Oral Vaccination. Clin. Vaccine Immunol. 2011, 18, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Hong, Q.; Qian, P.; Li, X.M.; Yu, X.L.; Chen, H.C. A recombinant pseudorabies virus co-expressing capsid proteins precursor P1-2A of FMDV and VP2 protein of porcine parvovirus: A trivalent vaccine candidate. Biotechnol. Lett. 2007, 29, 1677–1683. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.; Hassard, L.; Clark, E.; Harding, J.; Allan, G.; Willson, P.; Strokappe, J.; Martin, K.; McNeilly, F.; Meehan, B. Isolation of circovirus from lesions of pigs with postweaning multisystemic wasting syndrome. Can. Vet. J. 1998, 39, 44–51. [Google Scholar] [PubMed]
- Krakowka, S.; Ellis, J.A.; Meehan, B.; Kennedy, S.; McNeilly, F.; Allan, G. Viral wasting syndrome of swine: Experimental reproduction of postweaning multisystemic wasting syndrome in gnotobiotic swine by coinfection with porcine circovirus 2 and porcine parvovirus. Vet. Pathol. 2000, 37, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.; Larour, G.; Le Diguerher, G.; Eveno, E.; Jolly, J.P.; Blanchard, P.; Oger, A.; Le Dimna, M.; Jestin, A.; Madec, F. Risk factors for porcine post-weaning multisystemic wasting syndrome (PMWS) in 149 French farrow-to-finish herds. Prev. Vet. Med. 2003, 61, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Allan, G.M.; Kennedy, S.; McNeilly, F.; Foster, J.C.; Ellis, J.A.; Krakowka, S.J.; Meehan, B.M.; Adair, B.M. Experimental reproduction of severe wasting disease by co-infection of pigs with porcine circovirus and porcine parvovirus. J. Comp. Pathol. 1999, 121, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Chae, C. Distribution of porcine parvovirus in porcine circovirus 2-infected pigs with postweaning multisystemic wasting syndrome as shown by in-situ hybridization. J. Comp. Pathol. 2000, 123, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.A.; Bratanich, A.; Clark, E.G.; Allan, G.; Meehan, B.; Haines, D.M.; Harding, J.; West, K.H.; Krakowka, S.; Konoby, C.; et al. Coinfection by porcine circoviruses and porcine parvovirus in pigs with naturally acquired postweaning multisystemic wasting syndrome. J. Vet. Diagn. Investig. 2000, 12, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.; Moffett, D.; McNeilly, F.; Meehan, B.; Ellis, J.; Krakowka, S.; Allan, G.M. Reproduction of lesions of postweaning multisystemic wasting syndrome by infection of conventional pigs with porcine circovirus type 2 alone or in combination with porcine parvovirus. J. Comp. Pathol. 2000, 122, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.; Lee, Y.H.; Ahn, K.K.; Kim, B.; Chae, C. Reproduction of postweaning multisystemic wasting syndrome in pigs by prenatal porcine circovirus 2 infection and postnatal porcine parvovirus infection or immunostimulation. Vet. Pathol. 2008, 45, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Ostanello, F.; Caprioli, A.; di Francesco, A.; Battilani, M.; Sala, G.; Sarli, G.; Mandrioli, L.; McNeilly, F.; Allan, G.M.; Prosperi, S. Experimental infection of 3-week-old conventional colostrum-fed pigs with porcine circovirus type 2 and porcine parvovirus. Vet. Microbiol. 2005, 108, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.; Shin, J.H.; Chae, C. Colostral transmission of porcine circovirus 2 (PCV-2): Reproduction of post-weaning multisystemic wasting syndrome in pigs fed milk from PCV-2-infected sows with post-natal porcine parvovirus infection or immunostimulation. J. Gen. Virol. 2010, 91, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Grau-Roma, L.; Stockmarr, A.; Kristensen, C.S.; Enoe, C.; Lopez-Soria, S.; Nofrarias, M.; Bille-Hansen, V.; Hjulsager, C.K.; Sibila, M.; Jorsal, S.E.; et al. Infectious risk factors for individual postweaning multisystemic wasting syndrome (PMWS) development in pigs from affected farms in Spain and Denmark. Res. Vet. Sci. 2012, 93, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.E.; Nauwynck, H.J.; McNeilly, F.; Allan, G.M.; Pensaert, M.B. Porcine circovirus 2 infection in swine foetuses inoculated at different stages of gestation. Vet. Microbiol. 2001, 83, 169–176. [Google Scholar] [CrossRef]
- Joo, H.S.; Donaldson-Wood, C.R.; Johnson, R.H. A standardised haemagglutination inhibition test for porcine parvovirus antibody. Aust. Vet. J. 1976, 52, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Jeoung, H.Y.; Lim, S.I.; Kim, J.J.; Cho, Y.Y.; Kim, Y.K.; Song, J.Y.; Hyun, B.H.; An, D.J. Serological prevalence of viral agents that induce reproductive failure in South Korean wild boar. BMC Vet. Res. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Phuong, C.T.B.; Le, H.T.M. Prevalence of Antibodies to Porcine Parvovirus in Swine in Hanoi and Its Vicinity. In Proceedings of the Conference on Agriculture Development in the Context of International Integration: Opportunities and Challenges, Hanoi, Vietnam, 7–8 December 2016; p. 101. [Google Scholar]
- Qing, L.; Lv, J.; Li, H.; Tan, Y.; Hao, H.; Chen, Z.; Zhao, J.; Chen, H. The recombinant nonstructural polyprotein NS1 of porcine parvovirus (PPV) as diagnostic antigen in ELISA to differentiate infected from vaccinated pigs. Vet. Res. Commun. 2006, 30, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Song, C.P.; Zhu, C.; Zhang, C.F.; Cui, S.J. Detection of porcine parvovirus using a taqman-based real-time pcr with primers and probe designed for the NS1 gene. Virol. J. 2010, 7. [Google Scholar] [CrossRef] [PubMed]
- Streck, A.F.; Hergemoller, F.; Ruster, D.; Speck, S.; Truyen, U. A TaqMan qPCR for quantitation of Ungulate protoparvovirus 1 validated in several matrices. J. Virol. Methods 2015, 218, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.Q.; Cai, X.Q.; Lin, Z.X.; Li, X.L.; Yue, Q.Y.; Li, R.; Zhu, X.Q. Rapid and specific detection of porcine parvovirus using real-time PCR and High Resolution Melting (HRM) analysis. BMC Vet. Res. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.T.; Zhang, J.; Yang, S.H.; Ma, L.N.; Ma, Y.P.; Liu, X.T.; Cai, X.P.; Zhang, Y.G.; Liu, Y.S. Rapid detection of porcine parvovirus DNA by sensitive loop-mediated isothermal amplification. J. Virol. Methods 2009, 158, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Wang, Z.; Ma, X.; Liu, J.; Cui, S. A sensitive and specific nanoparticle-assisted PCR assay for rapid detection of porcine parvovirus. Lett. Appl. Microbiol. 2014, 58, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Qin, X.D.; Zhang, W.; Li, Y.M.; Zhang, Z.D. Rapid and specific detection of porcine parvovirus by isothermal recombinase polymerase amplification assays. Mol. Cell. Probes 2016, 30, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.G.; Chen, G.D.; Huang, Y.; Ding, L.; Li, Z.C.; Chang, C.D.; Wang, C.Y.; Tong, D.W.; Liu, H.J. Development of multiplex PCR for simultaneous detection of six swine DNA and RNA viruses. J. Virol. Methods 2012, 183, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.L.; Wang, Y.B.; Li, M.F.; Chen, H.Y.; Guo, X.P.; Geng, J.W.; Wang, Z.Y.; Wei, Z.Y.; Cui, B.A. Simultaneous detection of porcine parvovirus and porcine circovirus type 2 by duplex real-time PCR and amplicon melting curve analysis using SYBR Green. J. Virol. Methods 2013, 187, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.B.; Wu, H.G.; Jiang, Y.H.; Opriessnig, T.; Zheng, X.W.; Mo, Y.C.; Yang, Z.Q. Development of an EvaGreen-based multiplex real-time PCR assay with melting curve analysis for simultaneous detection and differentiation of six viral pathogens of porcine reproductive and respiratory disorder. J. Virol. Methods 2014, 208, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.G.; Rao, P.B.; Jiang, Y.H.; Opriessnig, T.; Yang, Z.Q. A sensitive multiplex real-time PCR panel for rapid diagnosis of viruses associated with porcine respiratory and reproductive disorders. Mol. Cell. Probes 2014, 28, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.Y.; Liu, Z.J.; Wang, W.C.; Tang, D.Y.; Liang, H.Y.; Liu, Z. Establishment and application of a multiplex PCR for rapid and simultaneous detection of six viruses in swine. J. Virol. Methods 2014, 208, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.X.; Xie, Z.X.; Xie, L.J.; Deng, X.W.; Xie, Z.Q.; Luo, S.S.; Liu, J.B.; Pang, Y.S.; Khan, M.I. Simultaneous detection of eight swine reproductive and respiratory pathogens using a novel GeXP analyser-based multiplex PCR assay. J. Virol. Methods 2015, 224, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Lin, X.Y.; Nie, F.P.; Yang, Z.X.; Yao, X.P.; Li, G.L.; Wu, X.L.; Ren, M.S.; Wang, Y. Simultaneous typing of seven porcine pathogens by multiplex PCR with a GeXP analyser. J. Virol. Methods 2016, 232, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Heldt, C.L.; Hernandez, R.; Mudiganti, U.; Gurgel, P.V.; Brown, D.T.; Carbonell, R.G. A colorimetric assay for viral agents that produce cytopathic effects. J. Virol. Methods 2006, 135, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Siegl, G.; Hallauer, C.; Novak, A. Paroviruses as contaminants of permanent human cell lines. IV. Multiplication of KBSH-virus in KB-cells. Arch. Gesamte Virusforsch. 1971, 36, 351–362. [Google Scholar] [CrossRef]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mészáros, I.; Olasz, F.; Cságola, A.; Tijssen, P.; Zádori, Z. Biology of Porcine Parvovirus (Ungulate parvovirus 1). Viruses 2017, 9, 393. https://doi.org/10.3390/v9120393
Mészáros I, Olasz F, Cságola A, Tijssen P, Zádori Z. Biology of Porcine Parvovirus (Ungulate parvovirus 1). Viruses. 2017; 9(12):393. https://doi.org/10.3390/v9120393
Chicago/Turabian StyleMészáros, István, Ferenc Olasz, Attila Cságola, Peter Tijssen, and Zoltán Zádori. 2017. "Biology of Porcine Parvovirus (Ungulate parvovirus 1)" Viruses 9, no. 12: 393. https://doi.org/10.3390/v9120393
APA StyleMészáros, I., Olasz, F., Cságola, A., Tijssen, P., & Zádori, Z. (2017). Biology of Porcine Parvovirus (Ungulate parvovirus 1). Viruses, 9(12), 393. https://doi.org/10.3390/v9120393