The Regulation of Translation in Alphavirus-Infected Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
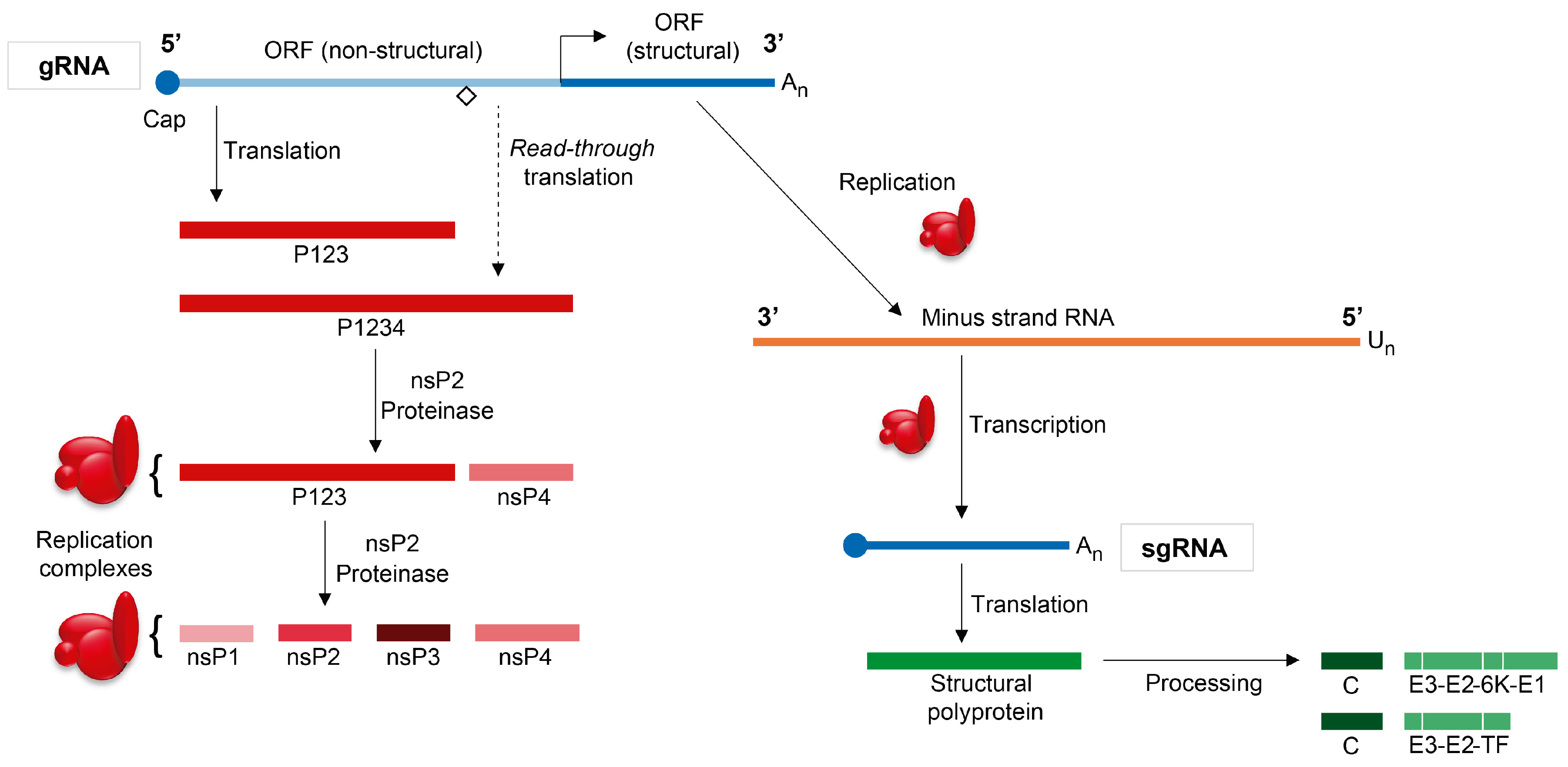
2. Overview of the Sindbis Virus Life Cycle
3. Inhibition of Host Translation by SINV Infection
3.1. Mechanisms of Inhibition of Cellular Protein Synthesis by SINV Infection
3.2. Involvement of nsP2 in Host Translation Shut-Off
3.3. Redistribution of Cellular Proteins between the Nucleus and Cytoplasm. A Proposal for the Mechanism of Cellular Translation Shut-Off
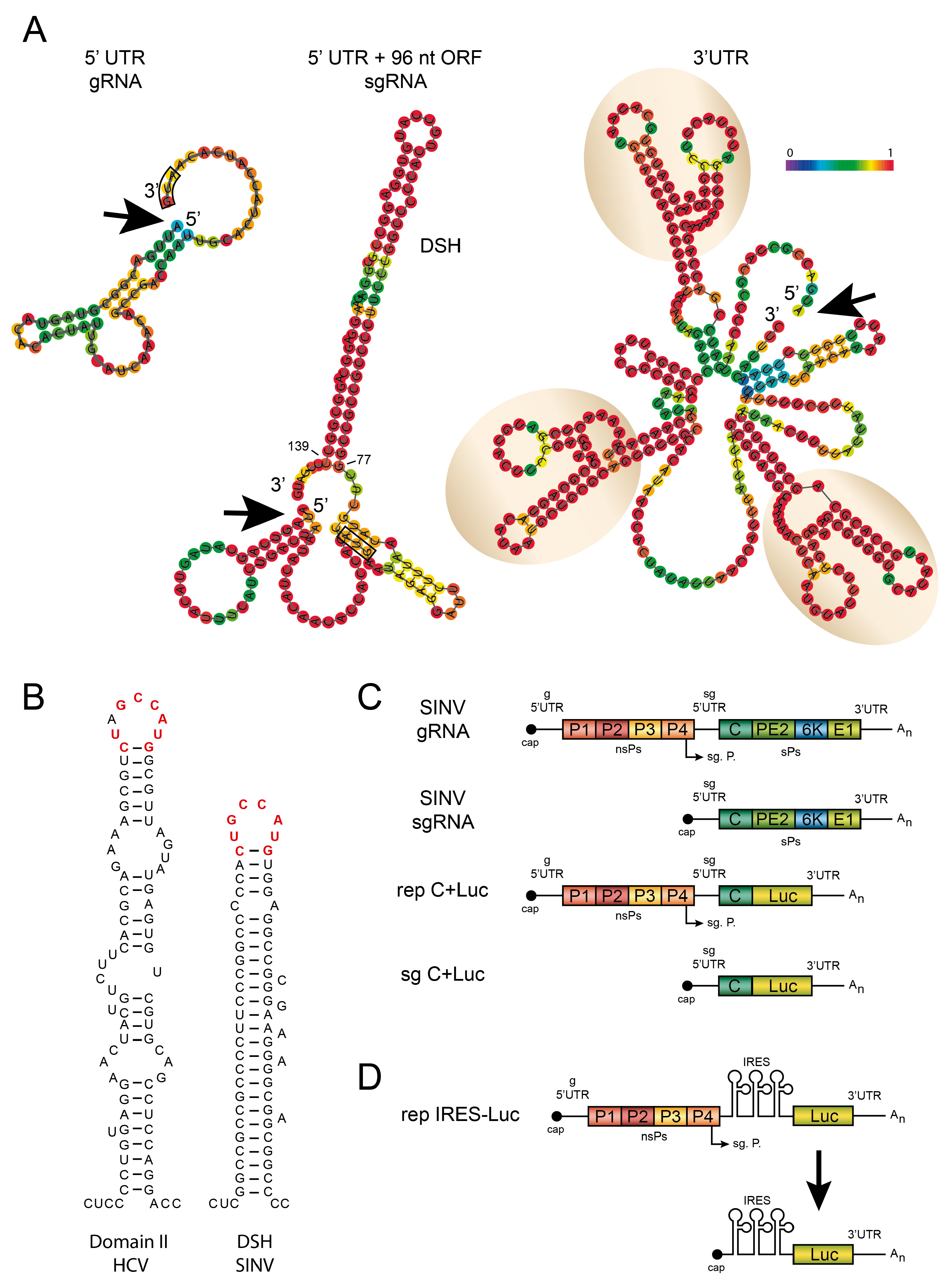
4. Structure of SINV sgRNA
4.1. The 5′-UTR of SINV sgRNA
4.2. The Hairpin Structure in the Coding Region of sgRNA
4.3. The 3′-UTR of SINV sgRNA
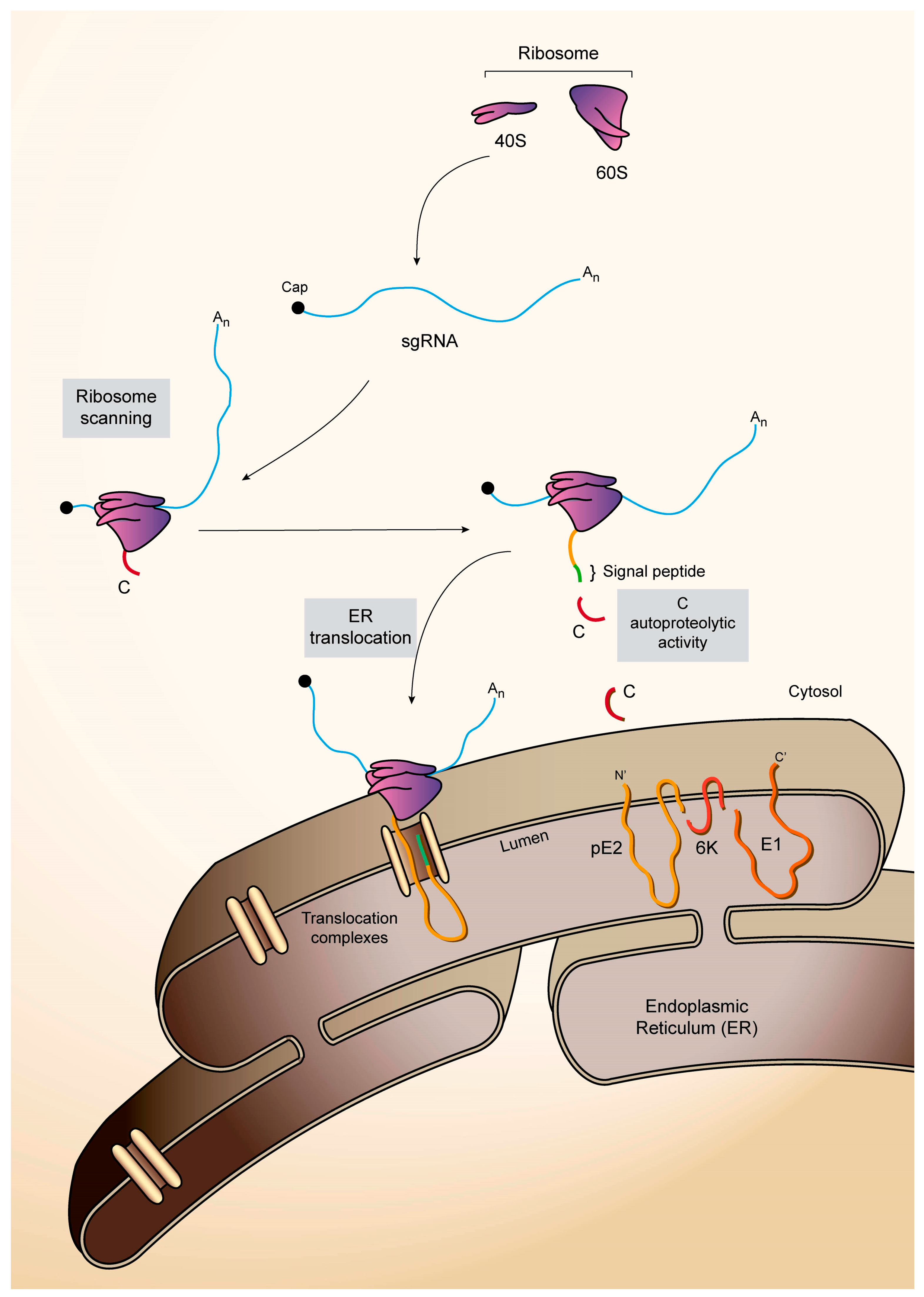
5. Mechanism of SINV sgRNA Translation
5.1. Protein Synthesis Directed by sgRNA without an Intact eIF4F Complex
5.2. Translation without eIF2
5.3. Mechanism of sgRNA Translation. Comparison with Cellular mRNAs
6. Translation of SINV sgRNA Bearing IRES Elements
6.1. The Variety of Internal Ribosome Entry Site Elements
6.2. IRES-Driven Translation in Alphavirus Replicons
7. Concluding Remarks and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Griffin, D.E. Alphaviruses. In Fields Virology; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1023–1068. [Google Scholar]
- Forrester, N.L.; Palacios, G.; Tesh, R.B.; Savji, N.; Guzman, H.; Sherman, M.; Weaver, S.C.; Lipkin, W.I. Genome-scale phylogeny of the alphavirus genus suggests a marine origin. J. Virol. 2012, 86, 2729–2738. [Google Scholar] [CrossRef] [PubMed]
- Nasar, F.; Palacios, G.; Gorchakov, R.V.; Guzman, H.; da Rosa, A.P.; Savji, N.; Popov, V.L.; Sherman, M.B.; Lipkin, W.I.; Tesh, R.B.; et al. Eilat virus, a unique alphavirus with host range restricted to insects by RNA replication. Proc. Natl. Acad. Sci. USA 2012, 109, 14622–14627. [Google Scholar] [CrossRef] [PubMed]
- Novella, I.S.; Presloid, J.B.; Smith, S.D.; Wilke, C.O. Specific and nonspecific host adaptation during arboviral experimental evolution. J. Mol. Microbiol. Biotechnol. 2011, 21, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Winegar, R.; Manger, I.D.; Forrester, N.L. Alphaviruses: Population genetics and determinants of emergence. Antivir. Res. 2012, 94, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Karpf, A.R.; Blake, J.M.; Brown, D.T. Characterization of the infection of Aedes albopictus cell clones by Sindbis virus. Virus Res. 1997, 50, 1–13. [Google Scholar] [CrossRef]
- Mudiganti, U.; Hernandez, R.; Ferreira, D.; Brown, D.T. Sindbis virus infection of two model insect cell systems—A comparative study. Virus Res. 2006, 122, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Suzme, R.; Tseng, J.C.; Levin, B.; Ibrahim, S.; Meruelo, D.; Pellicer, A. Sindbis viral vectors target hematopoietic malignant cells. Cancer Gene Ther. 2012, 19, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Tassetto, M.; Kunitomi, M.; Andino, R. Circulating Immune Cells Mediate a Systemic RNAi-Based Adaptive Antiviral Response in Drosophila. Cell 2017, 169, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Fuller, S.D. The T = 4 envelope of Sindbis virus is organized by interactions with a complementary T = 3 capsid. Cell 1987, 48, 923–934. [Google Scholar] [CrossRef]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [PubMed]
- Wang, K.S.; Kuhn, R.J.; Strauss, E.G.; Ou, S.; Strauss, J.H. High-affinity laminin receptor is a receptor for Sindbis virus in mammalian cells. J. Virol. 1992, 66, 4992–5001. [Google Scholar] [PubMed]
- Klimstra, W.B.; Nangle, E.M.; Smith, M.S.; Yurochko, A.D.; Ryman, K.D. DC-SIGN and L-SIGN can act as attachment receptors for alphaviruses and distinguish between mosquito cell- and mammalian cell-derived viruses. J. Virol. 2003, 77, 12022–12032. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.P.; Hanna, S.L.; Spiridigliozzi, A.; Wannissorn, N.; Beiting, D.P.; Ross, S.R.; Hardy, R.W.; Bambina, S.A.; Heise, M.T.; Cherry, S. Natural resistance-associated macrophage protein is a cellular receptor for sindbis virus in both insect and mammalian hosts. Cell Host Microbe 2011, 10, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.; Carrasco, L. Involvement of the vacuolar H+-ATPase in animal virus entry. J. Gen. Virol. 1994, 75 Pt 10, 2595–2606. [Google Scholar] [CrossRef] [PubMed]
- Mayor, S.; Pagano, R.E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.Y.; Ng, M.M.; Chu, J.J. Replication of alphaviruses: A review on the entry process of alphaviruses into cells. Adv. Virol. 2011, 2011, 249640. [Google Scholar] [CrossRef] [PubMed]
- Vancini, R.; Wang, G.; Ferreira, D.; Hernandez, R.; Brown, D.T. Alphavirus genome delivery occurs directly at the plasma membrane in a time- and temperature-dependent process. J. Virol. 2013, 87, 4352–4359. [Google Scholar] [CrossRef] [PubMed]
- Sokoloski, K.J.; Nease, L.M.; May, N.A.; Gebhart, N.N.; Jones, C.E.; Morrison, T.E.; Hardy, R.W. Identification of Interactions between Sindbis Virus Capsid Protein and Cytoplasmic vRNA as Novel Virulence Determinants. PLoS Pathog. 2017, 13, e1006473. [Google Scholar] [CrossRef] [PubMed]
- Berlanga, J.J.; Ventoso, I.; Harding, H.P.; Deng, J.; Ron, D.; Sonenberg, N.; Carrasco, L.; de Haro, C. Antiviral effect of the mammalian translation initiation factor 2α kinase GCN2 against RNA viruses. EMBO J. 2006, 25, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Shin, G.; Yost, S.A.; Miller, M.T.; Elrod, E.J.; Grakoui, A.; Marcotrigiano, J. Structural and functional insights into alphavirus polyprotein processing and pathogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 16534–16539. [Google Scholar] [CrossRef] [PubMed]
- Sokoloski, K.J.; Haist, K.C.; Morrison, T.E.; Mukhopadhyay, S.; Hardy, R.W. Noncapped Alphavirus Genomic RNAs and Their Role during Infection. J. Virol. 2015, 89, 6080–6092. [Google Scholar] [CrossRef] [PubMed]
- De Groot, R.J.; Hardy, W.R.; Shirako, Y.; Strauss, J.H. Cleavage-site preferences of Sindbis virus polyproteins containing the non-structural proteinase. Evidence for temporal regulation of polyprotein processing in vivo. EMBO J. 1990, 9, 2631–2638. [Google Scholar] [PubMed]
- Laakkonen, P.; Ahola, T.; Kaariainen, L. The effects of palmitoylation on membrane association of Semliki forest virus RNA capping enzyme. J. Biol. Chem. 1996, 271, 28567–28571. [Google Scholar] [CrossRef] [PubMed]
- Ahola, T.; Lampio, A.; Auvinen, P.; Kaariainen, L. Semliki Forest virus mRNA capping enzyme requires association with anionic membrane phospholipids for activity. EMBO J. 1999, 18, 3164–3172. [Google Scholar] [CrossRef] [PubMed]
- Ahola, T.; Kujala, P.; Tuittila, M.; Blom, T.; Laakkonen, P.; Hinkkanen, A.; Auvinen, P. Effects of palmitoylation of replicase protein nsP1 on alphavirus infection. J. Virol. 2000, 74, 6725–6733. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Rice, C.M. The signal for translational readthrough of a UGA codon in Sindbis virus RNA involves a single cytidine residue immediately downstream of the termination codon. J. Virol. 1993, 67, 5062–5067. [Google Scholar] [PubMed]
- Rupp, J.C.; Sokoloski, K.J.; Gebhart, N.N.; Hardy, R.W. Alphavirus RNA synthesis and non-structural protein functions. J. Gen. Virol. 2015, 96, 2483–2500. [Google Scholar] [CrossRef] [PubMed]
- Shirako, Y.; Strauss, E.G.; Strauss, J.H. Suppressor mutations that allow sindbis virus RNA polymerase to function with nonaromatic amino acids at the N-terminus: Evidence for interaction between nsP1 and nsP4 in minus-strand RNA synthesis. Virology 2000, 276, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Fata, C.L.; Sawicki, S.G.; Sawicki, D.L. Modification of Asn374 of nsP1 suppresses a Sindbis virus nsP4 minus-strand polymerase mutant. J. Virol. 2002, 76, 8641–8649. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Durbin, R.; Huang, H.V.; Rice, C.M.; Stollar, V. Association of the Sindbis virus RNA methyltransferase activity with the nonstructural protein nsP1. Virology 1989, 170, 385–391. [Google Scholar] [CrossRef]
- Ahola, T.; Laakkonen, P.; Vihinen, H.; Kaariainen, L. Critical residues of Semliki Forest virus RNA capping enzyme involved in methyltransferase and guanylyltransferase-like activities. J. Virol. 1997, 71, 392–397. [Google Scholar] [PubMed]
- Peranen, J.; Laakkonen, P.; Hyvonen, M.; Kaariainen, L. The alphavirus replicase protein nsP1 is membrane-associated and has affinity to endocytic organelles. Virology 1995, 208, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Salonen, A.; Vasiljeva, L.; Merits, A.; Magden, J.; Jokitalo, E.; Kaariainen, L. Properly folded nonstructural polyprotein directs the Semliki Forest virus replication complex to the endosomal compartment. J. Virol. 2003, 77, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.T.; White, M.A.; Watowich, S.J. The crystal structure of the Venezuelan equine encephalitis alphavirus nsP2 protease. Structure 2006, 14, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Gomez de Cedron, M.; Ehsani, N.; Mikkola, M.L.; Garcia, J.A.; Kaariainen, L. RNA helicase activity of Semliki Forest virus replicase protein NSP2. FEBS Lett. 1999, 448, 19–22. [Google Scholar] [CrossRef]
- Garmashova, N.; Gorchakov, R.; Frolova, E.; Frolov, I. Sindbis virus nonstructural protein nsP2 is cytotoxic and inhibits cellular transcription. J. Virol. 2006, 80, 5686–5696. [Google Scholar] [CrossRef] [PubMed]
- Breakwell, L.; Dosenovic, P.; Karlsson Hedestam, G.B.; D’Amato, M.; Liljestrom, P.; Fazakerley, J.; McInerney, G.M. Semliki Forest virus nonstructural protein 2 is involved in suppression of the type I interferon response. J. Virol. 2007, 81, 8677–8684. [Google Scholar] [CrossRef] [PubMed]
- Rikkonen, M.; Peranen, J.; Kaariainen, L. Nuclear targeting of Semliki Forest virus nsP2. Arch. Virol. Suppl. 1994, 9, 369–377. [Google Scholar] [PubMed]
- Akhrymuk, I.; Kulemzin, S.V.; Frolova, E.I. Evasion of the innate immune response: The Old World alphavirus nsP2 protein induces rapid degradation of Rpb1, a catalytic subunit of RNA polymerase II. J. Virol. 2012, 86, 7180–7191. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Gorbalenya, A.E.; Purdy, M.A.; Rozanov, M.N.; Reyes, G.R.; Bradley, D.W. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: Delineation of an additional group of positive-strand RNA plant and animal viruses. Proc. Natl. Acad. Sci. USA 1992, 89, 8259–8263. [Google Scholar] [CrossRef] [PubMed]
- Vihinen, H.; Ahola, T.; Tuittila, M.; Merits, A.; Kaariainen, L. Elimination of phosphorylation sites of Semliki Forest virus replicase protein nsP3. J. Biol. Chem. 2001, 276, 5745–5752. [Google Scholar] [CrossRef] [PubMed]
- Lulla, A.; Lulla, V.; Merits, A. Macromolecular assembly-driven processing of the 2/3 cleavage site in the alphavirus replicase polyprotein. J. Virol. 2012, 86, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Panas, M.D.; Varjak, M.; Lulla, A.; Eng, K.E.; Merits, A.; Karlsson Hedestam, G.B.; McInerney, G.M. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol. Biol. Cell 2012, 23, 4701–4712. [Google Scholar] [CrossRef] [PubMed]
- Fros, J.J.; Domeradzka, N.E.; Baggen, J.; Geertsema, C.; Flipse, J.; Vlak, J.M.; Pijlman, G.P. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J. Virol. 2012, 86, 10873–10879. [Google Scholar] [CrossRef] [PubMed]
- Rubach, J.K.; Wasik, B.R.; Rupp, J.C.; Kuhn, R.J.; Hardy, R.W.; Smith, J.L. Characterization of purified Sindbis virus nsP4 RNA-dependent RNA polymerase activity in vitro. Virology 2009, 384, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Pietila, M.K.; Hellstrom, K.; Ahola, T. Alphavirus polymerase and RNA replication. Virus Res. 2017, 234, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Shirako, Y.; Strauss, J.H. Regulation of Sindbis virus RNA replication: Uncleaved P123 and nsP4 function in minus-strand RNA synthesis, whereas cleaved products from P123 are required for efficient plus-strand RNA synthesis. J. Virol. 1994, 68, 1874–1885. [Google Scholar] [PubMed]
- Lemm, J.A.; Rumenapf, T.; Strauss, E.G.; Strauss, J.H.; Rice, C.M. Polypeptide requirements for assembly of functional Sindbis virus replication complexes: A model for the temporal regulation of minus- and plus-strand RNA synthesis. EMBO J. 1994, 13, 2925–2934. [Google Scholar] [PubMed]
- LaStarza, M.W.; Lemm, J.A.; Rice, C.M. Genetic analysis of the nsP3 region of Sindbis virus: Evidence for roles in minus-strand and subgenomic RNA synthesis. J. Virol. 1994, 68, 5781–5791. [Google Scholar] [PubMed]
- Kujala, P.; Ikaheimonen, A.; Ehsani, N.; Vihinen, H.; Auvinen, P.; Kaariainen, L. Biogenesis of the Semliki Forest virus RNA replication complex. J. Virol. 2001, 75, 3873–3884. [Google Scholar] [CrossRef] [PubMed]
- Kallio, K.; Hellstrom, K.; Balistreri, G.; Spuul, P.; Jokitalo, E.; Ahola, T. Template RNA length determines the size of replication complex spherules for Semliki Forest virus. J. Virol. 2013, 87, 9125–9134. [Google Scholar] [CrossRef] [PubMed]
- Kallio, K.; Hellstrom, K.; Jokitalo, E.; Ahola, T. RNA Replication and Membrane Modification Require the Same Functions of Alphavirus Nonstructural Proteins. J. Virol. 2015, 90, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.; Irurzun, A.; Carrasco, L. Action of brefeldin A on translation in Semliki Forest virus-infected HeLa cells and cells doubly infected with poliovirus. J. Gen. Virol. 1994, 75, 2197–2203. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, L.; Guinea, R.; Irurzun, A.; Barco, A. Effects of viral replication on cellular membrane metabolism and function. In Molecular Biology of Picornaviruses; Wimmer, E., Semler, B.L., Eds.; ASM Press: Washington, DC, USA, 2002; pp. 337–354. [Google Scholar]
- Hellstrom, K.; Kallio, K.; Utt, A.; Quirin, T.; Jokitalo, E.; Merits, A.; Ahola, T. Partially Uncleaved Alphavirus Replicase Forms Spherule Structures in the Presence and Absence of RNA Template. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Varjak, M.; Saul, S.; Arike, L.; Lulla, A.; Peil, L.; Merits, A. Magnetic fractionation and proteomic dissection of cellular organelles occupied by the late replication complexes of Semliki Forest virus. J. Virol. 2013, 87, 10295–10312. [Google Scholar] [CrossRef] [PubMed]
- Cristea, I.M.; Rozjabek, H.; Molloy, K.R.; Karki, S.; White, L.L.; Rice, C.M.; Rout, M.P.; Chait, B.T.; MacDonald, M.R. Host factors associated with the Sindbis virus RNA-dependent RNA polymerase: Role for G3BP1 and G3BP2 in virus replication. J. Virol. 2010, 84, 6720–6732. [Google Scholar] [CrossRef] [PubMed]
- Sariola, M.; Saraste, J.; Kuismanen, E. Communication of post-Golgi elements with early endocytic pathway: Regulation of endoproteolytic cleavage of Semliki Forest virus p62 precursor. J. Cell Sci. 1995, 108 Pt 6, 2465–2475. [Google Scholar] [PubMed]
- Firth, A.E.; Chung, B.Y.; Fleeton, M.N.; Atkins, J.F. Discovery of frameshifting in Alphavirus 6K resolves a 20-year enigma. Virol. J. 2008, 5, 108. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.Y.; Firth, A.E.; Atkins, J.F. Frameshifting in alphaviruses: A diversity of 3′ stimulatory structures. J. Mol. Biol. 2010, 397, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Jose, J.; Snyder, J.E.; Kuhn, R.J. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 2009, 4, 837–856. [Google Scholar] [CrossRef] [PubMed]
- Gaedigk-Nitschko, K.; Schlesinger, M.J. The Sindbis virus 6K protein can be detected in virions and is acylated with fatty acids. Virology 1990, 175, 274–281. [Google Scholar] [CrossRef]
- Sanz, M.A.; Perez, L.; Carrasco, L. Semliki Forest virus 6K protein modifies membrane permeability after inducible expression in Escherichia coli cells. J. Biol. Chem. 1994, 269, 12106–12110. [Google Scholar] [PubMed]
- Sanz, M.A.; Madan, V.; Carrasco, L.; Nieva, J.L. Interfacial domains in Sindbis virus 6K protein. Detection and functional characterization. J. Biol. Chem. 2003, 278, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, J.; Mukhopadhyay, S. Disentangling the Frames, the State of Research on the Alphavirus 6K and TF Proteins. Viruses 2017, 9, 228. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Carrasco, L. Sindbis virus variant with a deletion in the 6K gene shows defects in glycoprotein processing and trafficking: Lack of complementation by a wild-type 6K gene in trans. J. Virol. 2001, 75, 7778–7784. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.; Madan, V.; Nieva, J.L.; Carrasco, L. The Alphavirus 6K Protein. In Viral Membrane Proteins: Structure, Function, and Drug Design; Fisher, W., Ed.; Kluwer Academic/Plenum Plublishers: New York, NY, USA, 2005; pp. 233–244. [Google Scholar]
- Melton, J.V.; Ewart, G.D.; Weir, R.C.; Board, P.G.; Lee, E.; Gage, P.W. Alphavirus 6K proteins form ion channels. J. Biol. Chem. 2002, 277, 46923–46931. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.E.; Kulcsar, K.A.; Schultz, K.L.; Riley, C.P.; Neary, J.T.; Marr, S.; Jose, J.; Griffin, D.E.; Kuhn, R.J. Functional characterization of the alphavirus TF protein. J. Virol. 2013, 87, 8511–8523. [Google Scholar] [CrossRef] [PubMed]
- Bushell, M.; Sarnow, P. Hijacking the translation apparatus by RNA viruses. J. Cell Biol. 2002, 158, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Fros, J.J.; Pijlman, G.P. Alphavirus Infection: Host Cell Shut-Off and Inhibition of Antiviral Responses. Viruses 2016, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Moreno, M.; Sanz, M.A.; Pelletier, J.; Carrasco, L. Requirements for eIF4A and eIF2 during translation of Sindbis virus subgenomic mRNA in vertebrate and invertebrate host cells. Cell. Microbiol. 2013, 15, 823–840. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Garcia-Moreno, M.; Carrasco, L. Inhibition of host protein synthesis by Sindbis virus: Correlation with viral RNA replication and release of nuclear proteins to the cytoplasm. Cell. Microbiol. 2015, 17, 520–541. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Raught, B.; Sonenberg, N. eIF4 initiation factors: Effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 1999, 68, 913–963. [Google Scholar] [CrossRef] [PubMed]
- Parsyan, A.; Svitkin, Y.; Shahbazian, D.; Gkogkas, C.; Lasko, P.; Merrick, W.C.; Sonenberg, N. mRNA helicases: The tacticians of translational control. Nat. Rev. Mol. Cell Biol. 2011, 12, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Structural features in eukaryotic mRNAs that modulate the initiation of translation. J. Biol. Chem. 1991, 266, 19867–19870. [Google Scholar] [PubMed]
- Komar, A.A.; Mazumder, B.; Merrick, W.C. A new framework for understanding IRES-mediated translation. Gene 2012, 502, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Niepmann, M. Internal translation initiation of picornaviruses and hepatitis C virus. Biochim. Biophys. Acta 2009, 1789, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Moreno, M.; Sanz, M.A.; Carrasco, L. Initiation codon selection is accomplished by a scanning mechanism without crucial initiation factors in Sindbis virus subgenomic mRNA. RNA 2015, 21, 93–112. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. eIF2 and the control of cell physiology. Semin. Cell Dev. Biol. 2005, 16, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2α kinases: Their structures and functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Koromilas, A.E. Roles of the translation initiation factor eIF2α serine 51 phosphorylation in cancer formation and treatment. Biochim. Biophys. Acta 2015, 1849, 871–880. [Google Scholar] [CrossRef] [PubMed]
- McInerney, G.M.; Kedersha, N.L.; Kaufman, R.J.; Anderson, P.; Liljestrom, P. Importance of eIF2α phosphorylation and stress granule assembly in alphavirus translation regulation. Mol. Biol. Cell 2005, 16, 3753–3763. [Google Scholar] [CrossRef] [PubMed]
- Ventoso, I.; Sanz, M.A.; Molina, S.; Berlanga, J.J.; Carrasco, L.; Esteban, M. Translational resistance of late alphavirus mRNA to eIF2α phosphorylation: A strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev. 2006, 20, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Castello, A.; Ventoso, I.; Berlanga, J.J.; Carrasco, L. Dual mechanism for the translation of subgenomic mRNA from Sindbis virus in infected and uninfected cells. PLoS ONE 2009, 4, e4772. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Redondo, N.; Garcia-Moreno, M.; Carrasco, L. Phosphorylation of eIF2α is responsible for the failure of the picornavirus internal ribosome entry site to direct translation from Sindbis virus replicons. J. Gen. Virol. 2013, 94, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Gorchakov, R.; Frolova, E.; Williams, B.R.; Rice, C.M.; Frolov, I. PKR-dependent and -independent mechanisms are involved in translational shutoff during Sindbis virus infection. J. Virol. 2004, 78, 8455–8467. [Google Scholar] [CrossRef] [PubMed]
- Berglund, P.; Finzi, D.; Bennink, J.R.; Yewdell, J.W. Viral alteration of cellular translational machinery increases defective ribosomal products. J. Virol. 2007, 81, 7220–7229. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Burnham, A.J.; Gebhart, N.N.; Sokoloski, K.J.; Hardy, R.W. Role for subgenomic mRNA in host translation inhibition during Sindbis virus infection of mammalian cells. Virology 2013, 441, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Frolov, I.; Schlesinger, S. Comparison of the effects of Sindbis virus and Sindbis virus replicons on host cell protein synthesis and cytopathogenicity in BHK cells. J. Virol. 1994, 68, 1721–1727. [Google Scholar] [PubMed]
- Sanz, M.A.; Castello, A.; Carrasco, L. Viral translation is coupled to transcription in Sindbis virus-infected cells. J. Virol. 2007, 81, 7061–7068. [Google Scholar] [CrossRef] [PubMed]
- Garry, R.F. Sindbis virus-induced inhibition of protein synthesis is partially reversed by medium containing an elevated potassium concentration. J. Gen. Virol. 1994, 75, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Garry, R.F.; Bishop, J.M.; Parker, S.; Westbrook, K.; Lewis, G.; Waite, M.R. Na+ and K+ concentrations and the regulation of protein synthesis in Sindbis virus-infected chick cells. Virology 1979, 96, 108–120. [Google Scholar] [CrossRef]
- Carrasco, L. Membrane leakiness after viral infection and a new approach to the development of antiviral agents. Nature 1978, 272, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Contreras, A.; Carrasco, L. Selective inhibition of protein synthesis in virus-infected mammalian cells. J. Virol. 1979, 29, 114–122. [Google Scholar] [PubMed]
- Frolov, I.; Garmashova, N.; Atasheva, S.; Frolova, E.I. Random Insertion Mutagenesis of Sindbis Virus Nonstructural Protein 2 and Selection of Variants Incapable of Downregulating Cellular Transcription. J. Virol. 2009, 83, 9031–9044. [Google Scholar] [CrossRef] [PubMed]
- Frolov, I.; Agapov, E.; Hoffman, T.A., Jr.; Pragai, B.M.; Lippa, M.; Schlesinger, S.; Rice, C.M. Selection of RNA replicons capable of persistent noncytopathic replication in mammalian cells. J. Virol. 1999, 73, 3854–3865. [Google Scholar] [PubMed]
- Castello, A.; Alvarez, E.; Carrasco, L. The multifaceted poliovirus 2A protease: Regulation of gene expression by picornavirus proteases. J. Biomed. Biotechnol. 2011, 2011, 369648. [Google Scholar] [CrossRef] [PubMed]
- McCormick, C.; Khaperskyy, D.A. Translation inhibition and stress granules in the antiviral immune response. Nat. Rev. Immunol. 2017, 17, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Gui, H.; Lu, C.W.; Adams, S.; Stollar, V.; Li, M.L. hnRNP A1 interacts with the genomic and subgenomic RNA promoters of Sindbis virus and is required for the synthesis of G and SG RNA. J. Biomed. Sci. 2010, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Dickson, A.M.; Anderson, J.R.; Barnhart, M.D.; Sokoloski, K.J.; Oko, L.; Opyrchal, M.; Galanis, E.; Wilusz, C.J.; Morrison, T.E.; Wilusz, J. Dephosphorylation of HuR protein during alphavirus infection is associated with HuR relocalization to the cytoplasm. J. Biol. Chem. 2012, 287, 36229–36238. [Google Scholar] [CrossRef] [PubMed]
- Burnham, A.J.; Gong, L.; Hardy, R.W. Heterogeneous nuclear ribonuclear protein K interacts with Sindbis virus nonstructural proteins and viral subgenomic mRNA. Virology 2007, 367, 212–221. [Google Scholar] [CrossRef] [PubMed]
- LaPointe, A.T.; Gebhart, N.N.; Meller, M.E.; Hardy, R.W.; Sokoloski, K.J. The Identification and Characterization of Sindbis Virus RNA: Host Protein Interactions. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sokoloski, K.J.; Dickson, A.M.; Chaskey, E.L.; Garneau, N.L.; Wilusz, C.J.; Wilusz, J. Sindbis virus usurps the cellular HuR protein to stabilize its transcripts and promote productive infections in mammalian and mosquito cells. Cell Host Microbe 2010, 8, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, M.D.; Moon, S.L.; Emch, A.W.; Wilusz, C.J.; Wilusz, J. Changes in cellular mRNA stability, splicing, and polyadenylation through HuR protein sequestration by a cytoplasmic RNA virus. Cell Rep. 2013, 5, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Izquierdo, J.M.; Welnowska, E.; Carrasco, L. RNA nuclear export is blocked by poliovirus 2A protease and is concomitant with nucleoporin cleavage. J. Cell Sci. 2009, 122, 3799–3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, N.; Katikaneni, P.; Skern, T.; Gustin, K.E. Differential targeting of nuclear pore complex proteins in poliovirus-infected cells. J. Virol. 2008, 82, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Park, N.; Skern, T.; Gustin, K.E. Specific cleavage of the nuclear pore complex protein Nup62 by a viral protease. J. Biol. Chem. 2010, 285, 28796–28805. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, E.; Castello, A.; Carrasco, L.; Izquierdo, J.M. Poliovirus 2A protease triggers a selective nucleo-cytoplasmic redistribution of splicing factors to regulate alternative pre-mRNA splicing. PLoS ONE 2013, 8, e73723. [Google Scholar]
- Alvarez, E.; Castello, A.; Carrasco, L.; Izquierdo, J.M. Alternative splicing, a new target to block cellular gene expression by poliovirus 2A protease. Biochem. Biophys. Res. Commun. 2011, 414, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Hyde, J.L.; Chen, R.; Trobaugh, D.W.; Diamond, M.S.; Weaver, S.C.; Klimstra, W.B.; Wilusz, J. The 5′ and 3′ ends of alphavirus RNAs—Non-coding is not non-functional. Virus Res. 2015, 206, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Sanz, M.A.; Molina, S.; Carrasco, L. Translation of Sindbis virus 26S mRNA does not require intact eukariotic initiation factor 4G. J. Mol. Biol. 2006, 355, 942–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frolov, I.; Schlesinger, S. Translation of Sindbis virus mRNA: Effects of sequences downstream of the initiating codon. J. Virol. 1994, 68, 8111–8117. [Google Scholar] [PubMed]
- Frolov, I.; Schlesinger, S. Translation of Sindbis virus mRNA: Analysis of sequences downstream of the initiating AUG codon that enhance translation. J. Virol. 1996, 70, 1182–1190. [Google Scholar] [PubMed]
- Ventoso, I. Adaptive changes in alphavirus mRNA translation allowed colonization of vertebrate hosts. J. Virol. 2012, 86, 9484–9494. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Gonzalez Almela, E.; Carrasco, L. Translation of Sindbis Subgenomic mRNA is Independent of eIF2, eIF2A and eIF2D. Sci. Rep. 2017, 7, 43876. [Google Scholar] [CrossRef] [PubMed]
- Toribio, R.; Diaz-Lopez, I.; Boskovic, J.; Ventoso, I. An RNA trapping mechanism in Alphavirus mRNA promotes ribosome stalling and translation initiation. Nucleic Acids Res. 2016, 44, 4368–4380. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.W. The role of the 3′ terminus of the Sindbis virus genome in minus-strand initiation site selection. Virology 2006, 345, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.H.; Trent, D.W.; Strauss, J.H. The 3′-non-coding regions of alphavirus RNAs contain repeating sequences. J. Mol. Biol. 1982, 156, 719–730. [Google Scholar] [CrossRef]
- Pfeffer, M.; Kinney, R.M.; Kaaden, O.R. The alphavirus 3′-nontranslated region: Size heterogeneity and arrangement of repeated sequence elements. Virology 1998, 240, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Gritsun, T.S.; Gould, E.A. Direct repeats in the 3′ untranslated regions of mosquito-borne flaviviruses: Possible implications for virus transmission. J. Gen. Virol. 2006, 87, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Garneau, N.L.; Sokoloski, K.J.; Opyrchal, M.; Neff, C.P.; Wilusz, C.J.; Wilusz, J. The 3′ untranslated region of sindbis virus represses deadenylation of viral transcripts in mosquito and Mammalian cells. J. Virol. 2008, 82, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R.J.; Hong, Z.; Strauss, J.H. Mutagenesis of the 3′ nontranslated region of Sindbis virus RNA. J. Virol. 1990, 64, 1465–1476. [Google Scholar] [PubMed]
- Kuhn, R.J.; Griffin, D.E.; Zhang, H.; Niesters, H.G.; Strauss, J.H. Attenuation of Sindbis virus neurovirulence by using defined mutations in nontranslated regions of the genome RNA. J. Virol. 1992, 66, 7121–7127. [Google Scholar] [PubMed]
- Chen, R.; Wang, E.; Tsetsarkin, K.A.; Weaver, S.C. Chikungunya virus 3′ untranslated region: Adaptation to mosquitoes and a population bottleneck as major evolutionary forces. PLoS Pathog. 2013, 9, e1003591. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Moreno, M.; Sanz, M.A.; Carrasco, L. A Viral mRNA Motif at the 3′-Untranslated Region that Confers Translatability in a Cell-Specific Manner. Implications for Virus Evolution. Sci. Rep. 2016, 6, 19217. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, M.; Villoing, S.; Rimstad, E.; Nylund, A. Characterization of untranslated regions of the salmonid alphavirus 3 (SAV3) genome and construction of a SAV3 based replicon. Virol. J. 2009, 6, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLoughlin, M.F.; Graham, D.A. Alphavirus infections in salmonids—A review. J. Fish Dis. 2007, 30, 511–531. [Google Scholar] [CrossRef] [PubMed]
- Moriette, C.; Leberre, M.; Lamoureux, A.; Lai, T.L.; Bremont, M. Recovery of a recombinant salmonid alphavirus fully attenuated and protective for rainbow trout. J. Virol. 2006, 80, 4088–4098. [Google Scholar] [CrossRef] [PubMed]
- Weston, J.; Villoing, S.; Bremont, M.; Castric, J.; Pfeffer, M.; Jewhurst, V.; McLoughlin, M.; Rodseth, O.; Christie, K.E.; Koumans, J.; et al. Comparison of two aquatic alphaviruses, salmon pancreas disease virus and sleeping disease virus, by using genome sequence analysis, monoclonal reactivity, and cross-infection. J. Virol. 2002, 76, 6155–6163. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.E.; Steitz, J.A. Virus meets host microRNA: The destroyer, the booster, the hijacker. Mol. Cell. Biol. 2014, 34, 3780–3787. [Google Scholar] [CrossRef] [PubMed]
- Trobaugh, D.W.; Klimstra, W.B. MicroRNA Regulation of RNA Virus Replication and Pathogenesis. Trends Mol. Med. 2017, 23, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Trobaugh, D.W.; Gardner, C.L.; Sun, C.; Haddow, A.D.; Wang, E.; Chapnik, E.; Mildner, A.; Weaver, S.C.; Ryman, K.D.; Klimstra, W.B. RNA viruses can hijack vertebrate microRNAs to suppress innate immunity. Nature 2014, 506, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Trobaugh, D.W.; Klimstra, W.B. Alphaviruses suppress host immunity by preventing myeloid cell replication and antagonizing innate immune responses. Curr. Opin. Virol. 2017, 23, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Skalsky, R.L.; Kennedy, E.M.; Furuse, Y.; Whisnant, A.W.; Flores, O.; Schultz, K.L.; Putnam, N.; Barrows, N.J.; Sherry, B.; et al. Replication of many human viruses is refractory to inhibition by endogenous cellular microRNAs. J. Virol. 2014, 88, 8065–8076. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.S.; Schmid, S.; Aguado, L.C.; Sabin, L.R.; Yasunaga, A.; Shim, J.V.; Sachs, D.; Cherry, S.; tenOever, B.R. Drosha as an interferon-independent antiviral factor. Proc. Natl. Acad. Sci. USA 2014, 111, 7108–7113. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, J.; Han, Y.; Fan, X.; Ding, S.W. RNA interference functions as an antiviral immunity mechanism in mammals. Science 2013, 342, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.A.; Carrasco, L. Translation of capped viral mRNAs in poliovirus-infected HeLa cells. EMBO J. 1982, 1, 913–917. [Google Scholar] [PubMed]
- Castello, A.; Franco, D.; Moral-Lopez, P.; Berlanga, J.J.; Alvarez, E.; Wimmer, E.; Carrasco, L. HIV-1 protease inhibits Cap- and poly(A)-dependent translation upon eIF4GI and PABP cleavage. PLoS ONE 2009, 4, e7997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.S.; Kranzusch, P.J.; Doudna, J.A.; Cate, J.H. eIF3d is an mRNA cap-binding protein that is required for specialized translation initiation. Nature 2016, 536, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Skabkin, M.A.; Skabkina, O.V.; Dhote, V.; Komar, A.A.; Hellen, C.U.; Pestova, T.V. Activities of Ligatin and MCT-1/DENR in eukaryotic translation initiation and ribosomal recycling. Genes Dev. 2010, 24, 1787–1801. [Google Scholar] [CrossRef] [PubMed]
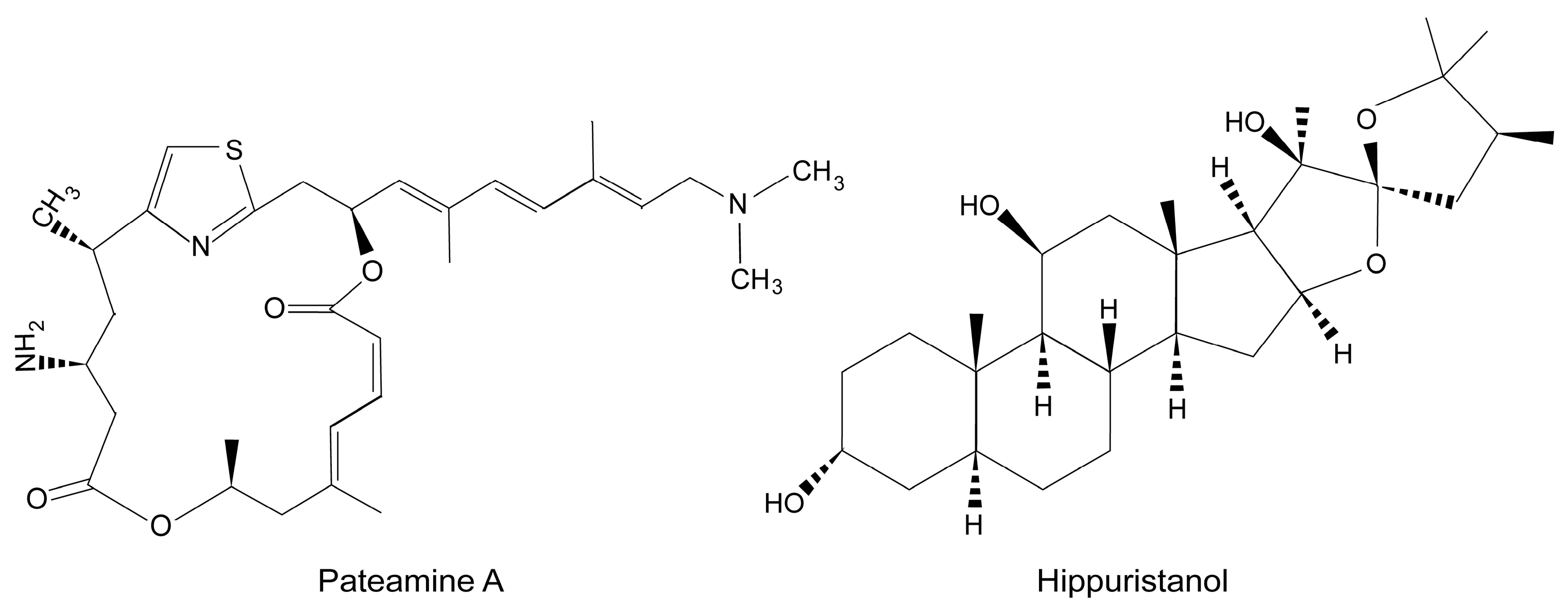
- Cencic, R.; Galicia-Vazquez, G.; Pelletier, J. Inhibitors of translation targeting eukaryotic translation initiation factor 4A. Methods Enzymol. 2012, 511, 437–461. [Google Scholar] [PubMed]
- Hood, K.A.; West, L.M.; Northcote, P.T.; Berridge, M.V.; Miller, J.H. Induction of apoptosis by the marine sponge (Mycale) metabolites, mycalamide A and pateamine. Apoptosis 2001, 6, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Low, W.K.; Dang, Y.; Schneider-Poetsch, T.; Shi, Z.; Choi, N.S.; Rzasa, R.M.; Shea, H.A.; Li, S.; Park, K.; Ma, G.; et al. Isolation and identification of eukaryotic initiation factor 4A as a molecular target for the marine natural product Pateamine A. Methods Enzymol. 2007, 431, 303–324. [Google Scholar] [PubMed]
- Bordeleau, M.E.; Mori, A.; Oberer, M.; Lindqvist, L.; Chard, L.S.; Higa, T.; Belsham, G.J.; Wagner, G.; Tanaka, J.; Pelletier, J. Functional characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat. Chem. Biol. 2006, 2, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Low, W.K.; Dang, Y.; Schneider-Poetsch, T.; Shi, Z.; Choi, N.S.; Merrick, W.C.; Romo, D.; Liu, J.O. Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol. Cell 2005, 20, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Kedersha, N.; Low, W.K.; Romo, D.; Gorospe, M.; Kaufman, R.; Anderson, P.; Liu, J.O. Eukaryotic initiation factor 2α-independent pathway of stress granule induction by the natural product pateamine A. J. Biol. Chem. 2006, 281, 32870–32878. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Almela, E.; Sanz, M.A.; Garcia-Moreno, M.; Northcote, P.; Pelletier, J.; Carrasco, L. Differential action of pateamine A on translation of genomic and subgenomic mRNAs from Sindbis virus. Virology 2015, 484, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, L.; Oberer, M.; Reibarkh, M.; Cencic, R.; Bordeleau, M.E.; Vogt, E.; Marintchev, A.; Tanaka, J.; Fagotto, F.; Altmann, M.; et al. Selective pharmacological targeting of a DEAD box RNA helicase. PLoS ONE 2008, 3, e1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoll, W.L.; Horton, L.E.; Komar, A.A.; Hensold, J.O.; Merrick, W.C. Characterization of mammalian eIF2A and identification of the yeast homolog. J. Biol. Chem. 2002, 277, 37079–37087. [Google Scholar] [CrossRef] [PubMed]
- Merrick, W.C.; Kemper, W.M.; Kantor, J.A.; Anderson, W.F. Purification and properties of rabbit reticulocyte protein synthesis elongation factor 2. J. Biol. Chem. 1975, 250, 2620–2625. [Google Scholar] [PubMed]
- Adams, S.L.; Safer, B.; Anderson, W.F.; Merrick, W.C. Eukaryotic initiation complex formation. Evidence for two distinct pathways. J. Biol. Chem. 1975, 250, 9083–9089. [Google Scholar] [PubMed]
- Liang, H.; He, S.; Yang, J.; Jia, X.; Wang, P.; Chen, X.; Zhang, Z.; Zou, X.; McNutt, M.A.; Shen, W.H.; et al. PTENα, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism. Cell Metab. 2014, 19, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Starck, S.R.; Tsai, J.C.; Chen, K.; Shodiya, M.; Wang, L.; Yahiro, K.; Martins-Green, M.; Shastri, N.; Walter, P. Translation from the 5′ untranslated region shapes the integrated stress response. Science 2016, 351. [Google Scholar] [CrossRef] [PubMed]
- Sendoel, A.; Dunn, J.G.; Rodriguez, E.H.; Naik, S.; Gomez, N.C.; Hurwitz, B.; Levorse, J.; Dill, B.D.; Schramek, D.; Molina, H.; et al. Translation from unconventional 5′ start sites drives tumour initiation. Nature 2017, 541, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Golovko, A.; Kojukhov, A.; Guan, B.J.; Morpurgo, B.; Merrick, W.C.; Mazumder, B.; Hatzoglou, M.; Komar, A.A. The eIF2A knockout mouse. Cell Cycle 2016, 15, 3115–3120. [Google Scholar] [CrossRef] [PubMed]
- Dmitriev, S.E.; Terenin, I.M.; Andreev, D.E.; Ivanov, P.A.; Dunaevsky, J.E.; Merrick, W.C.; Shatsky, I.N. GTP-independent tRNA delivery to the ribosomal P-site by a novel eukaryotic translation factor. J. Biol. Chem. 2010, 285, 26779–26787. [Google Scholar] [CrossRef] [PubMed]
- Marceau, C.D.; Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Brewer, S.M.; Fuchs, G.; Swaminathan, K.; Mata, M.A.; Elias, J.E.; Sarnow, P.; et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 2016, 535, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Boehringer, D.; Thermann, R.; Ostareck-Lederer, A.; Lewis, J.D.; Stark, H. Structure of the hepatitis C virus IRES bound to the human 80S ribosome: Remodeling of the HCV IRES. Structure 2005, 13, 1695–1706. [Google Scholar] [CrossRef] [PubMed]
- Locker, N.; Easton, L.E.; Lukavsky, P.J. HCV and CSFV IRES domain II mediate eIF2 release during 80S ribosome assembly. EMBO J. 2007, 26, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Lorsch, J.R.; Dever, T.E. Molecular view of 43 S complex formation and start site selection in eukaryotic translation initiation. J. Biol. Chem. 2010, 285, 21203–21207. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, A.G. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 2011, 75, 434–467. [Google Scholar] [CrossRef] [PubMed]
- Valasek, L.S. ‘Ribozoomin’—Translation initiation from the perspective of the ribosome-bound eukaryotic initiation factors (eIFs). Curr. Protein Pept. Sci. 2012, 13, 305–330. [Google Scholar] [CrossRef] [PubMed]
- Pestova, T.V.; Kolupaeva, V.G. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 2002, 16, 2906–2922. [Google Scholar] [CrossRef] [PubMed]
- Asano, K. Why is start codon selection so precise in eukaryotes? Translation 2014, 2, e28387. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Initiation of translation in prokaryotes and eukaryotes. Gene 1999, 234, 187–208. [Google Scholar] [CrossRef]
- Cheung, Y.N.; Maag, D.; Mitchell, S.F.; Fekete, C.A.; Algire, M.A.; Takacs, J.E.; Shirokikh, N.; Pestova, T.; Lorsch, J.R.; Hinnebusch, A.G. Dissociation of eIF1 from the 40S ribosomal subunit is a key step in start codon selection in vivo. Genes Dev. 2007, 21, 1217–1230. [Google Scholar] [CrossRef] [PubMed]
- Luna, R.E.; Arthanari, H.; Hiraishi, H.; Nanda, J.; Martin-Marcos, P.; Markus, M.A.; Akabayov, B.; Milbradt, A.G.; Luna, L.E.; Seo, H.C.; et al. The C-terminal domain of eukaryotic initiation factor 5 promotes start codon recognition by its dynamic interplay with eIF1 and eIF2β. Cell Rep. 2012, 1, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanda, J.S.; Saini, A.K.; Munoz, A.M.; Hinnebusch, A.G.; Lorsch, J.R. Coordinated movements of eukaryotic translation initiation factors eIF1, eIF1A, and eIF5 trigger phosphate release from eIF2 in response to start codon recognition by the ribosomal preinitiation complex. J. Biol. Chem. 2013, 288, 5316–5329. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.S.; Bai, X.C.; Murshudov, G.; Scheres, S.H.; Ramakrishnan, V. Initiation of translation by cricket paralysis virus IRES requires its translocation in the ribosome. Cell 2014, 157, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.; Savva, C.G.; Shin, B.S.; Dever, T.E.; Ramakrishnan, V.; Fernandez, I.S. Structural characterization of ribosome recruitment and translocation by type IV IRES. eLife 2016, 5, e13567. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Welnowska, E.; Redondo, N.; Carrasco, L. Translation driven by picornavirus IRES is hampered from Sindbis virus replicons: Rescue by poliovirus 2A protease. J. Mol. Biol. 2010, 402, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Kieft, J.S. Viral IRES RNA structures and ribosome interactions. Trends Biochem. Sci. 2008, 33, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Balvay, L.; Soto Rifo, R.; Ricci, E.P.; Decimo, D.; Ohlmann, T. Structural and functional diversity of viral IRESes. Biochim. Biophys. Acta 2009, 1789, 542–557. [Google Scholar] [CrossRef] [PubMed]
- Lozano, G.; Martinez-Salas, E. Structural insights into viral IRES-dependent translation mechanisms. Curr. Opin. Virol. 2015, 12, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Khawaja, A.; Vopalensky, V.; Pospisek, M. Understanding the potential of hepatitis C virus internal ribosome entry site domains to modulate translation initiation via their structure and function. Wiley Interdiscip. Rev. RNA 2015, 6, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.K. Internal initiation: IRES elements of picornaviruses and hepatitis c virus. Virus Res. 2006, 119, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, N.; Uchiumi, T. Functional analysis of structural motifs in dicistroviruses. Virus Res. 2009, 139, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Chen, C.J.; Shih, S.R. Regulation Mechanisms of Viral IRES-Driven Translation. Trends Microbiol. 2017, 25, 546–561. [Google Scholar] [CrossRef] [PubMed]
- Belsham, G.J. Divergent picornavirus IRES elements. Virus Res. 2009, 139, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Salas, E.; Francisco-Velilla, R.; Fernandez-Chamorro, J.; Lozano, G.; Diaz-Toledano, R. Picornavirus IRES elements: RNA structure and host protein interactions. Virus Res. 2015, 206, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Filbin, M.E.; Kieft, J.S. Toward a structural understanding of IRES RNA function. Curr. Opin. Struct. Biol. 2009, 19, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Lukavsky, P.J. Structure and function of HCV IRES domains. Virus Res. 2009, 139, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Butcher, S.E.; Jan, E. tRNA-mimicry in IRES-mediated translation and recoding. RNA Biol. 2016, 13, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Redondo, N.; Sanz, M.A.; Welnowska, E.; Carrasco, L. Translation without eIF2 promoted by poliovirus 2A protease. PLoS ONE 2011, 6, e25699. [Google Scholar] [CrossRef] [PubMed]
- Redondo, N.; Sanz, M.A.; Steinberger, J.; Skern, T.; Kusov, Y.; Carrasco, L. Translation directed by hepatitis A virus IRES in the absence of active eIF4F complex and eIF2. PLoS ONE 2012, 7, e52065. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Salas, E.; Lozano, G.; Fernandez-Chamorro, J.; Francisco-Velilla, R.; Galan, A.; Diaz, R. RNA-binding proteins impacting on internal initiation of translation. Int. J. Mol. Sci. 2013, 14, 21705–21726. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.G.; Grosely, R.; Petrov, A.N.; Puglisi, J.D. Dynamics of IRES-mediated translation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.E.; Carrasco, L. Eukaryotic viral protein synthesis. In ATP International Review of Science, Biochemistry Series II, Synthesis of Amino Acids and Proteins; Arnstein, H.V.R., Ed.; Butterworths: London, UK, 1978; Volume 18, pp. 261–311. [Google Scholar]
- Kamrud, K.I.; Custer, M.; Dudek, J.M.; Owens, G.; Alterson, K.D.; Lee, J.S.; Groebner, J.L.; Smith, J.F. Alphavirus replicon approach to promoterless analysis of IRES elements. Virology 2007, 360, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Plante, K.; Wang, E.; Partidos, C.D.; Weger, J.; Gorchakov, R.; Tsetsarkin, K.; Borland, E.M.; Powers, A.M.; Seymour, R.; Stinchcomb, D.T.; et al. Novel chikungunya vaccine candidate with an IRES-based attenuation and host range alteration mechanism. PLoS Pathog. 2011, 7, e1002142. [Google Scholar] [CrossRef] [PubMed]
- Erasmus, J.H.; Rossi, S.L.; Weaver, S.C. Development of Vaccines for Chikungunya Fever. J. Infect. Dis. 2016, 214, S488–S496. [Google Scholar] [CrossRef] [PubMed]
- Rausalu, K.; Iofik, A.; Ulper, L.; Karo-Astover, L.; Lulla, V.; Merits, A. Properties and use of novel replication-competent vectors based on Semliki Forest virus. Virol. J. 2009, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Welnowska, E.; Sanz, M.A.; Redondo, N.; Carrasco, L. Translation of viral mRNA without active eIF2: The case of picornaviruses. PLoS ONE 2011, 6, e22230. [Google Scholar] [CrossRef] [PubMed]
- Moral-Lopez, P.; Alvarez, E.; Redondo, N.; Skern, T.; Carrasco, L. L protease from foot and mouth disease virus confers eIF2-independent translation for mRNAs bearing picornavirus IRES. FEBS Lett. 2014, 588, 4053–4059. [Google Scholar] [CrossRef] [PubMed]
- Niepmann, M. Hepatitis C virus RNA translation. Curr. Top. Microbiol. Immunol. 2013, 369, 143–166. [Google Scholar] [PubMed]
- Hellen, C.U. IRES-induced conformational changes in the ribosome and the mechanism of translation initiation by internal ribosomal entry. Biochim. Biophys. Acta 2009, 1789, 558–570. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrasco, L.; Sanz, M.A.; González-Almela, E. The Regulation of Translation in Alphavirus-Infected Cells. Viruses 2018, 10, 70. https://doi.org/10.3390/v10020070
Carrasco L, Sanz MA, González-Almela E. The Regulation of Translation in Alphavirus-Infected Cells. Viruses. 2018; 10(2):70. https://doi.org/10.3390/v10020070
Chicago/Turabian StyleCarrasco, Luis, Miguel Angel Sanz, and Esther González-Almela. 2018. "The Regulation of Translation in Alphavirus-Infected Cells" Viruses 10, no. 2: 70. https://doi.org/10.3390/v10020070
APA StyleCarrasco, L., Sanz, M. A., & González-Almela, E. (2018). The Regulation of Translation in Alphavirus-Infected Cells. Viruses, 10(2), 70. https://doi.org/10.3390/v10020070