1. Introduction
Chikungunya virus (CHIKV) is a mosquito borne arthritogenic alphavirus responsible for numerous outbreaks of arthritic disease with significant human morbidity. Widely regarded as a nonfatal self-limiting disease, chikungunya gained global attention during the 2005–2006 Indian Ocean outbreak due to the severe and atypical nature of clinical symptoms [
1,
2]. Since then, CHIKV has spread rapidly in a relatively short period of time, most recently making incursions into the Americas where local transmission has been identified in over 40 countries or territories [
3].
Belonging to the
Togaviridae family, CHIKV is an arthritogenic alphavirus. Other arthritogenic alphaviruses include Sindbis virus (SINV), o’nyong nyong virus (ONNV), and Ross River virus. Encephalitic alphaviruses—such as Eastern, Western, and Venezuelan equine encephalitis viruses—are responsible for sporadic cases of human and equine neurological disease and are largely found in the western hemisphere. CHIKV, as with all alphaviruses, has a single strand positive-sense ~12-kb RNA genome. Genomic RNA serves as the mRNA for translation of four non-structural proteins (nsP1–4), which form the replicative enzyme complex responsible for viral genome replication, and transcription of a subgenomic RNA. The latter encodes five virus structural proteins (capsid protein, E3, E2, 6K/TF, and E1). The non-structural and structural proteins are each translated as polyprotein precursors that undergo proteolytic cleavage to form the mature viral proteins. The multifunctional capsid protein has a number of key roles. Through the activity of a serine protease catalytic site, the capsid protein cleaves itself from the nascent structural polyprotein. In a structural capacity, capsid protein specifically recognizes the packaging signals present in viral genomic RNA, allowing assembly of the nucleocapsid core [
4]. There are though functional differences between arthritogenic and encephalitic alphavirus capsid proteins. In mammalian cells, encephalitic alphavirus capsid proteins inhibit cellular transcription, while for arthritogenic viruses cellular transcription is antagonized by nsP2 [
5,
6]. The capsid protein of encephalitic alphaviruses is highly cytotoxic and this protein’s ability to shutdown host transcription is closely linked to an interaction with the nuclear pore complex [
5,
7].
Both arthritogenic and encephalitic alphavirus capsid proteins traffic to the mammalian host cell nucleus [
5,
8]. Disruption of capsid protein nuclear trafficking in encephalitic alphaviruses can impact on virulence and pathogenesis in vivo [
5]. Despite this, little is known of this importance of capsid protein nuclear trafficking in arthritogenic alphaviruses including CHIKV.
The presence of nuclear import and export signals in the capsid proteins of a number of alphaviruses, including CHIKV, have been documented [
7,
8,
9,
10,
11]. However, the regions within capsid protein responsible for nuclear export have been found to vary greatly between different alphaviral clades and species. Here, using site directed mutagenesis, we identify amino acid residues required for nuclear export and, using replicon systems, we show that mutating the nuclear export sequence of CHIKV capsid protein blocks host protein access to the nucleus.
2. Materials and Methods
2.1. Oligonucleotides, Plasmids, and Antibodies
Insertion of a PCR amplicon encoding the CHIKV capsid gene, from primers CHIKV-CAP-PCR XhoI F (GGCCCTCGAGAGTTCATCCCAACC) and CHIKV-CAP-PCR HindIII R (GCGCAAGCTTTACCACTCTTCGGCCC), into the XhoI-HindII sites in pEGFP-C1 generated pEGFP-CHIKV capsid. The mutations L51A and M53A within the CHIKV capsid protein encoding region were generated using overlapping PCR products. The primers CHIK-CAP-PCR XhoI F and ∆NES-L51A, M53A R (CGCGCgcTGTCgcTTTATTAACTGCTGAGATCAG) were used to generate the 5′ region of capsid containing the mutations at its 3′. While, the primers ∆NES-L51A, M53A F (AAAgcGACAgcGCGCGCGGTACCACAACAG), and CHIK-CAP-PCR HindIII R were used to generate the 3′ region of capsid encoding the mutations at its 5′. Following gel purification these PCR products were mixed and included in a PCR reaction with the primers CHIK-CAP-PCR XhoI F and CHIK-CAP-PCR HindIII R. The resultant amplicon was cloned into pEGFP-C1 between the XhoI and HindIII site to generate pEGFP-CHIKV capsid-∆NES.
The plasmid pSP6-ICRES1-ΔNES was generated by subcloning the AgeI-SfiI fragment containing the subgenomic promoter (SGP) and 5′ of the capsid gene from pSP6-ICRES1 (generated from the CHIKV strain LR2006_OPY1 and kindly provided by Andres Merits at the University of Tartu) into pCDNA.31(+) (Invitrogen, Carlsbad, CA, USA) to generate the pCDNA3.1(+)-SGP-CAP shuttle vector. The nucleic acid fragment encoding the capsid nuclear export sequence (NES) contained on a KpnI fragment was exchanged with that from the plasmid pEGFP-CHIKV capsid-ΔNES, described above. The AgeI-SflI fragment containing the ΔNES was returned to pSP6-ICRES1 to generate pSP6-ICRES1-ΔNES.
All CHIKV replicons encoding the capsid protein were constructed using pSP6-ChikRepl-SG2, kindly provided by Andres Merits at the University of Tartu. A NheI-SmaI fragment encoding the EGFP-capsid fusion (WT and ΔNES mutant) was removed from pEGFP-CHIKV-Capsid/pEGFP-CHIKV-Capsid ΔNES and cloned into pSP6-ChikRepl-SG2 using the unique AvrII and PmeI sites. This placed CHIKV capsid under the control of the subgenomic promoter of the replicon vector.
The CHIKV replicon encoding fusion protein consisting of four copies of mCherry protein harbouring the SV40 NLS was generated in three stages. Firstly, a PCR amplicon encoding mCherry was cloned into pSP6-CHKVRep-SG-ZsGreen to replace the ZsGreen gene using the AvrII and PmeI sites. Secondly, oligonucleotides encoding three copies of the SV40 NLS were synthesized and cloned at the 5′ of the mCherry gene using the SgrAI site located at the 3′ of mCherry and PmeI of the plasmid to generate SGP-AvrII-mCherry-SgrAI-3X NLS-PmeI. A control replicon lacking the 3XNLS was also generated.
All Venezuelan equine encephalitis virus (VEEV) capsid constructs were prepared as the CHKV EGFP-Capsid constructs. The VEEV capsid gene was synthesized by Dundee Cells Products (Dundee) based on the Trinidad Donkey/TC83 (GI:323714/GI:323714) vaccine strain and used as a template for all VEEV PCR reactions. The sequences of these and other constructs were verified by sequencing. Antibodies against capsid proteins were kindly provided by Roy Hall at The University of Queensland. Antibodies against nsP2 were kindly provided by Andres Merits at the University of Tartu.
2.2. Cell Culture and Virus Propagation
BHK-21 cells (Sigma-Aldrich, St. Louis, MO, USA) were propagated in Opti-MEM (Life Technologies, Carlsbad, CA, USA) supplemented with 3% Fetal bovine serum (FBS) (Bovogen, Melbourne, Australia). Infectious viruses were rescued by linearization of icDNA containing plasmids with NotI and in vitro transcription from the SP6 promoter using the AmpliCapTM SP6 High Yield Message Maker Kit (CELLSRIPT, Madison, WI, USA). Transcripts were electroporated into Vero cells and supernatants collected and titrated by plaque assay as described previously [
12]. Plasmid transfections were carried out with Lipofectamine
® 2000 (Life Technologies), according to the manufacturer’s protocol.
2.3. Immunofluorescence Microscopy
Cells were grown on polylysine-treated coverslips. For Leptomycin B treatment, 45 nM of Leptomycin B was added to cells for 30 min at 37 °C immediately prior to imaging. Cells were fixed in 4% paraformaldehyde and permeabilised in 1% Triton X-100. Cells were then blocked in 1% bovine serum albumin (BSA) made in PBS and incubated at 37 °C for 1 h. Primary antibodies were diluted 1:100 in 1% BSA and incubated with the cells for 1 h at 37 °C. Alexa Fluor 488 secondary antibody (Invitrogen) was diluted 1:500 in 1% BSA and incubated with the cells for 1 h at 37 °C. Coverslips were mounted in Vectorshield mounting medium (Vector Laboratories, Burlingame, CA, USA) and staining was visualized on an Olympus FluoView™ FV1000 confocal microscope.
4. Discussion
As positive-strand RNA viruses, alphaviruses replicate within the cytoplasm of host cells. Despite this, several alphaviruses target a number of viral proteins to the nucleus. The rationale for this is not immediately obvious, however, previous studies have highlighted host cell shut-off as one facet of capsid protein nuclear localization in encephalitic alphaviruses [
7]. Compartmentalization of viral proteins within subcellular structures of the host cell is therefore an important mechanism used by alphaviruses to augment protein function and optimize infectivity, particularly for a protein with multiple roles during infection such as CHIKV capsid protein.
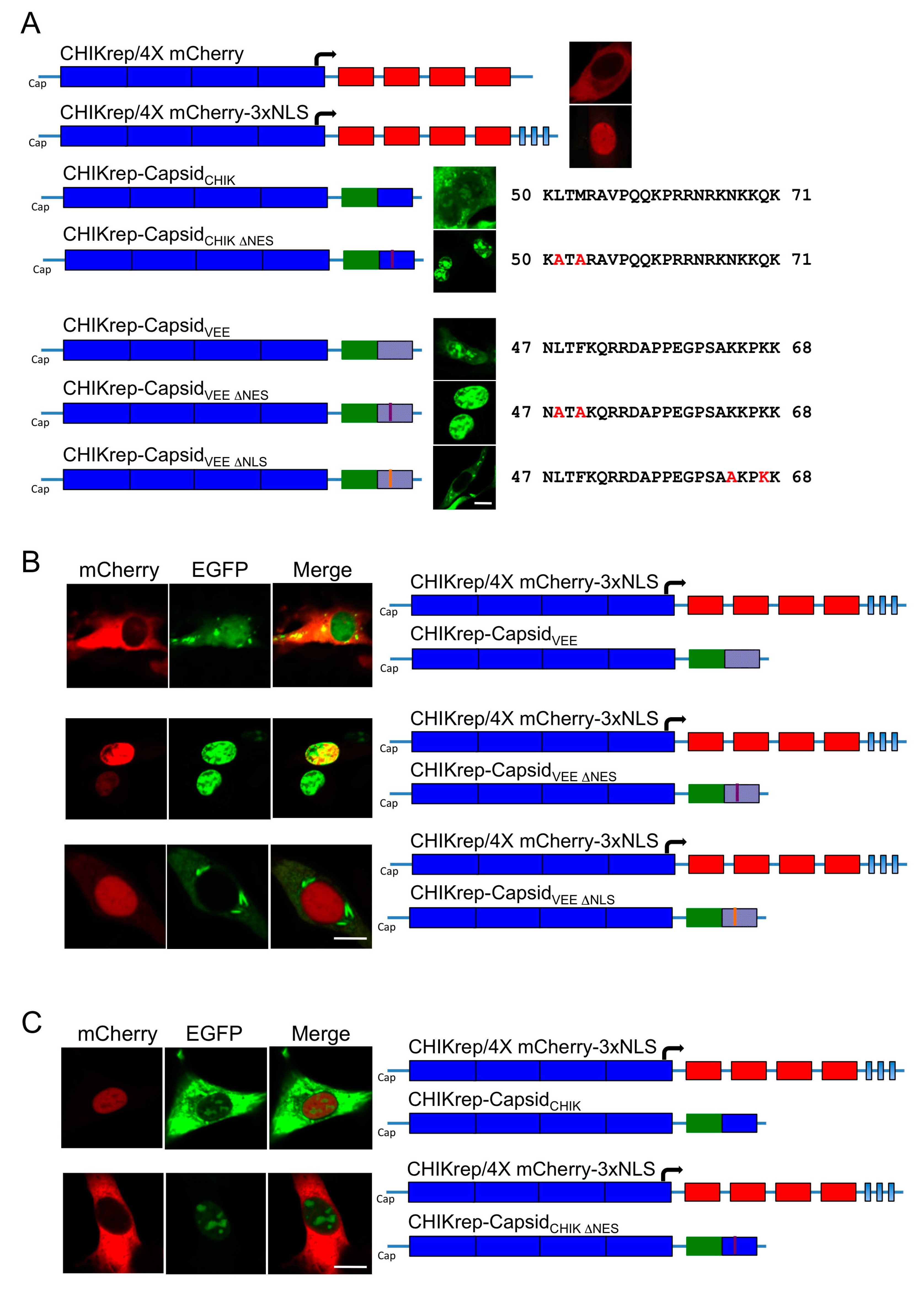
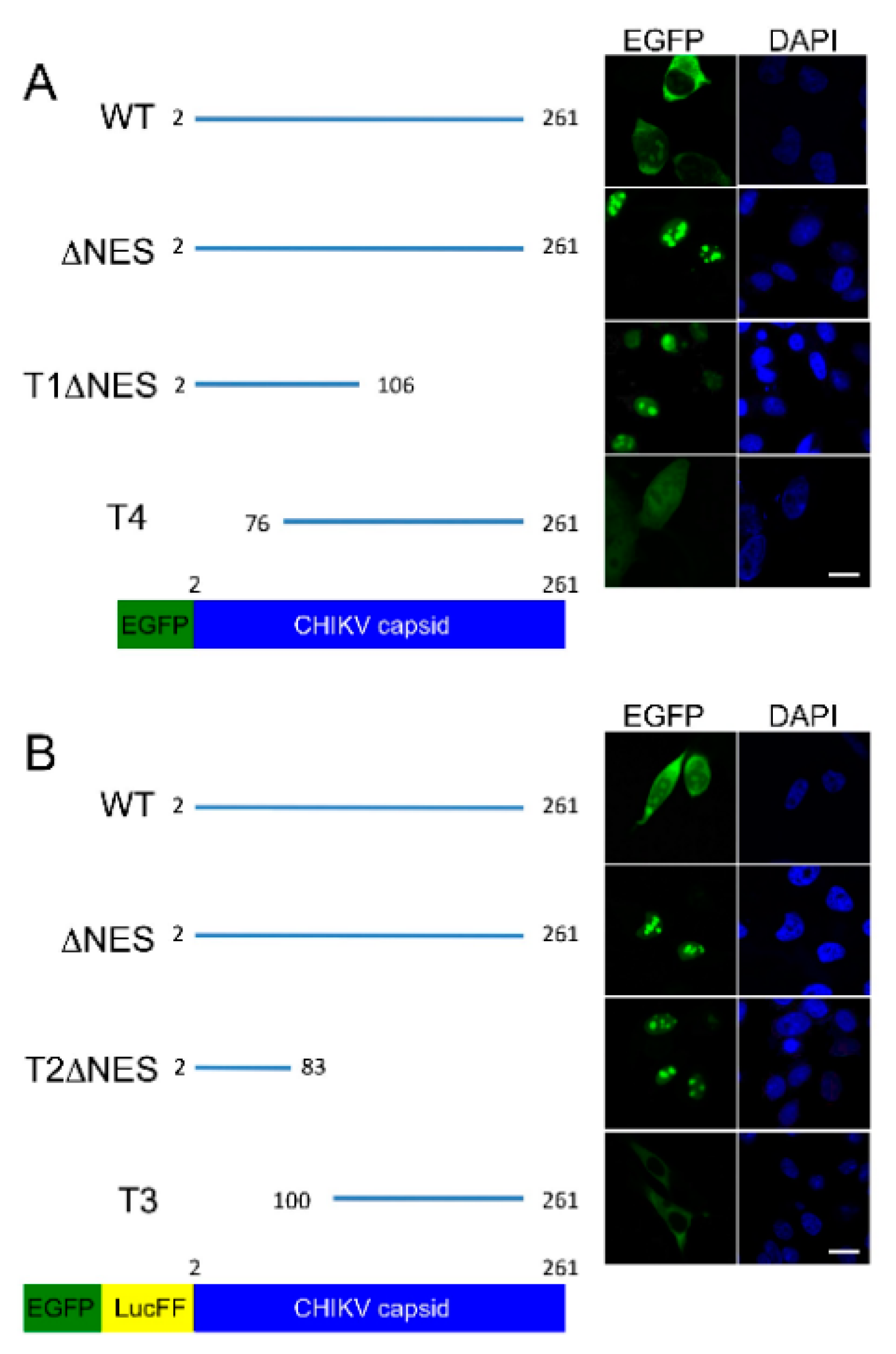
CHIKV capsid protein localizes to the cytoplasm and the nucleus in over expression studies. Our results outline a minimal NES, with homology to the NES of VEEV capsid protein, consisting of hydrophobic amino acid residues in the N terminal of the capsid protein. The identified NES is recognized as a classical, or prototypical, NES as it conforms to the traditional consensus Φ-X
2–3-Φ-X
2–3-Φ-X-Φ where Φ represents leucine, isoleucine, phenylalanine, valine, or methionine and X can be any amino acid. This NES differs from the one previously reported in CHIKV capsid protein, which describes a non-classical sequence towards the centre of the protein [
8]. It is therefore possible that CHIKV capsid contains two NESs that must both be intact for functional export of capsid from the nucleus, stressing the importance of capsid protein nuclear shuttling in CHIKV replication. Our results further delineate the nuclear localization signal (NLS) of CHIKV capsid to the N terminal 83 amino acids, which comprises a previously reported NLS between amino acids 60 and 100 [
8].
In accordance with previous studies, export of CHIKV capsid protein was CRM1 dependent, with capsid unable to exit the nucleus upon leptomycin B treatment. The active removal of capsid from the nucleus via a CRM1 mediated NES suggests that, in infected cells, capsid is actively exported from the nucleus and that this may be common to all alphavirus capsid proteins encoding this motif. Also, luciferase fusion constructs of CHIKV capsid, which would be excluded from the nucleus by passive diffusion due to their size, were observed in the nucleus confirming that capsid is actively transported to the nucleus. Given that capsid protein is observed predominantly in the cytoplasm of WT CHIKV infected cells, and that in CHIKV ΔNES infected cells capsid protein was observed in the nucleus, our results confirm that capsid actively and rapidly traverses the nucleus during infection.
Interestingly, in this study, mutation of CHIKV capsid protein NES blocked host cell nuclear import. Further studies will be required to understand the block to nuclear import caused when the ∆NES mutation is introduced into CHIKV capsid. Previous studies suggest the formation of a complex containing VEEV capsid is able to accumulate in the nuclear pore complex blocking nuclear import [
7]. Furthermore, the relative position and/or sequence between VEEV capsid NES and NLS was important in blocking the nuclear pore complex. Given that we have mapped a NLS in the N terminal 83 amino acids of CHIKV capsid it is possible that mutating the NES of CHIKV capsid permits the formation of a structure capable of blocking nuclear import, similar to that seen with WT VEEV capsid protein. Additionally, in CHIKV ∆NES infected cells, a high proportion of capsid protein was observed at the nuclear periphery indicative of an association with the nuclear pores, as seen with WT VEEV capsid protein [
5]. With an abundance of capsid protein found in the cytoplasm during infection, it also remains unclear whether all or just a proportion of capsid protein is able to traffic through the nucleus. The block to nuclear import may also reflect the fact that, in this replicon system, CHIKV capsid is expressed together with CHIKV nsP2, a protein shown to traffic to the nucleus in arthritogenic alphaviruses but that inefficiently translocates to the nucleus in encephalitic alphaviruses [
6,
14,
15]. Although the localization of nsP2 remained unchanged in CHIKV ∆NES infected cells, a block to nuclear import caused by CHIKV ∆NES capsid may represent an important yet undefined role for nsP2 in the trafficking of proteins, including capsid, through the nuclear membrane. The nuclear trafficking of alphaviral proteins warrants further investigation.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}


