Polyprotein Processing as a Determinant for in Vitro Activity of Semliki Forest Virus Replicase

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Plasmids, and Viruses
2.2. Metabolic Labeling with 3H-Uridine
2.3. Isolation of RCs from Infected or Transfected Cells
2.4. Purification of Membranes
2.5. IVRA, RNA Isolation, Agarose Gel Electrophoresis, and in-Gel Hybridization
2.6. Hybridization of 32P-labeled RNA Products with Capture Probes
2.7. 35S-Labeling and Protein Analyses
3. Results
3.1. Isolation of Active Replication Complexes from SFV-Infected Cells
3.2. Application of the In Vitro Replication Assay to the Trans-Replication System
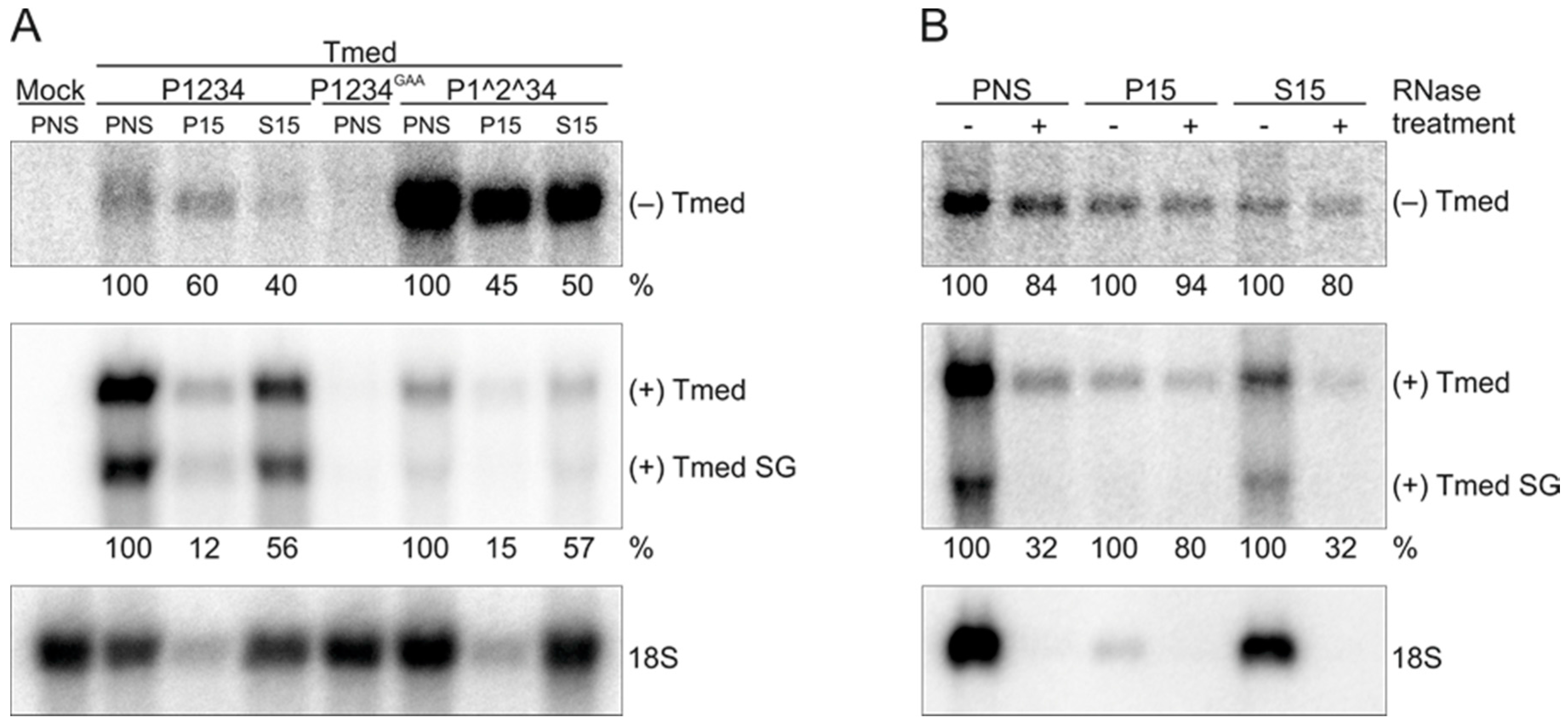
3.3. Polarity of SFV RNA Synthesized In Vitro
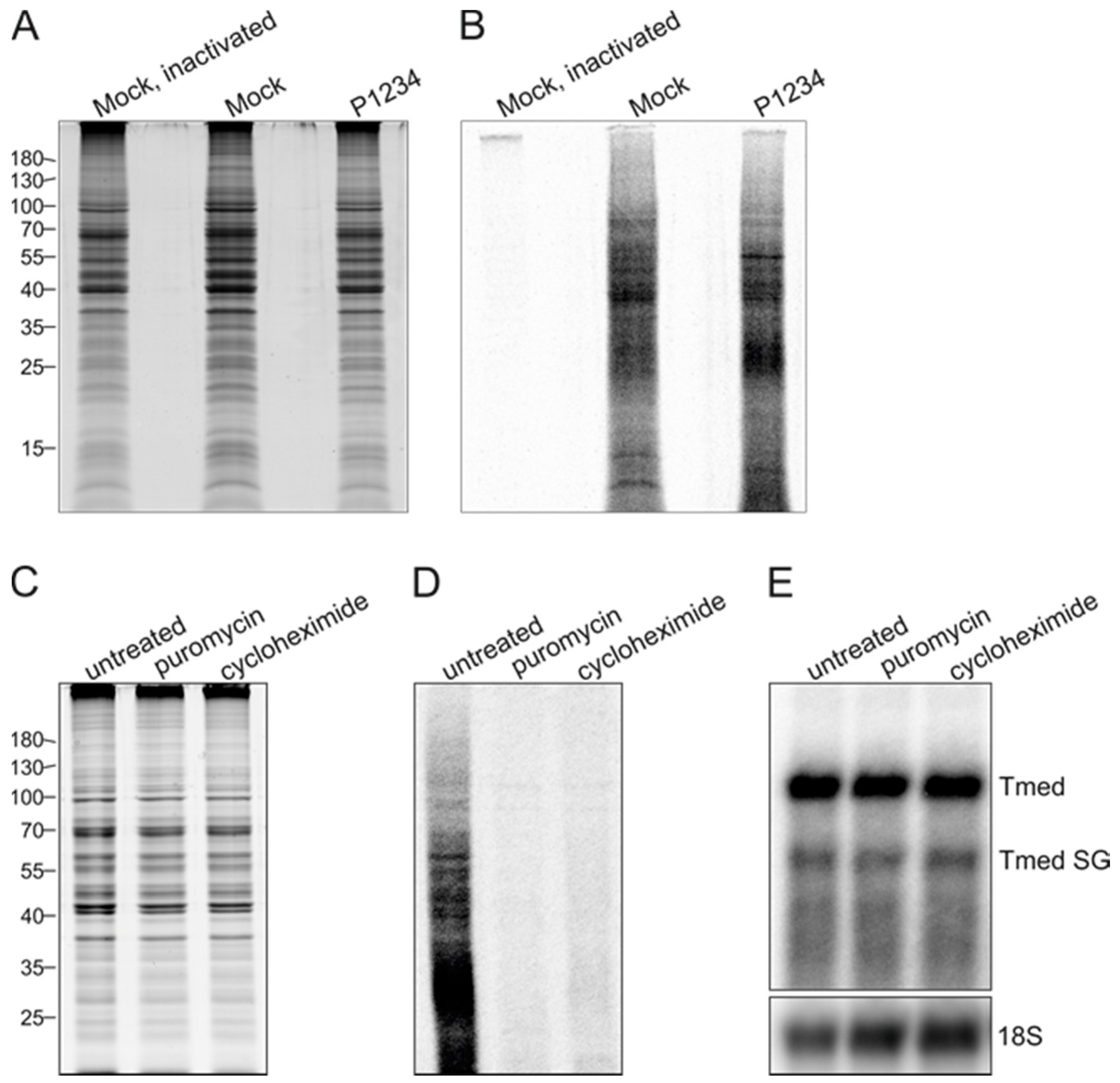
3.4. Effect of Ongoing Translation on In Vitro RNA Synthesizing Activity
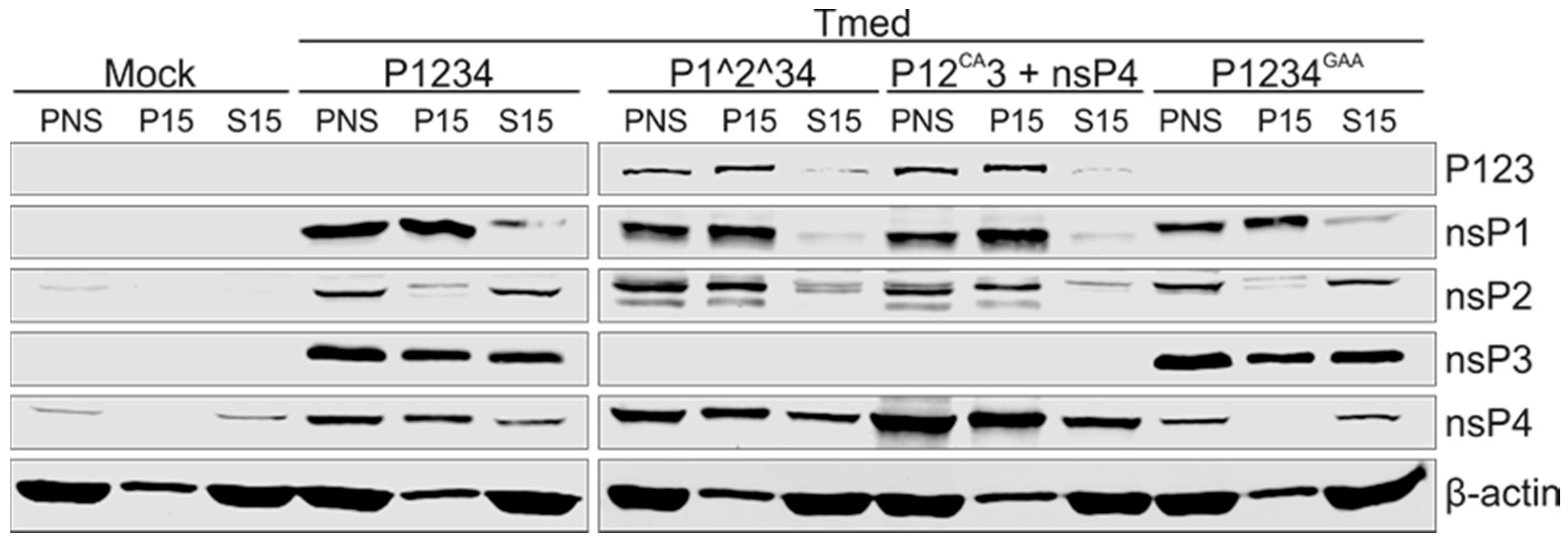
3.5. Purification of Replicase Protein-Containing Membranes and Addition of Exogenous Template RNA
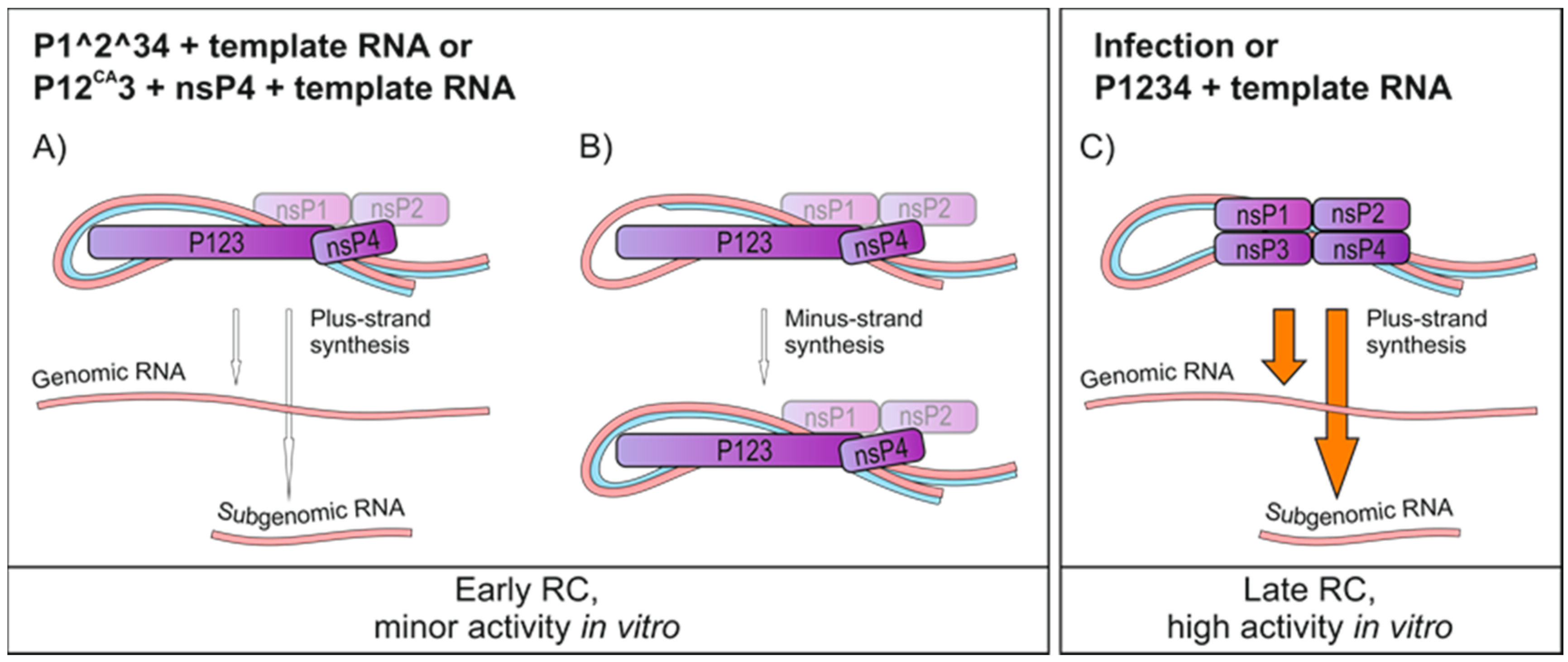
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. Virus Taxonomy: Ninth Report of the International Commitee on Taxonmy of Viruses; Elsevier Academic Press: London, UK, 2012; pp. 10–14. [Google Scholar]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. Origins and evolution of viruses of eukaryotes: The ultimate modularity. Virology 2015, 479, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Amraoui, F.; Failloux, A.B. Chikungunya: An unexpected emergence in Europe. Curr. Opin. Virol. 2016, 21, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Chen, R.; Weaver, S.C. Interspecies transmission and chikungunya virus emergence. Curr. Opin. Virol. 2016, 16, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [PubMed]
- Lemm, J.A.; Rice, C.M. Roles of nonstructural polyproteins and cleavage products in regulating Sindbis virus RNA replication and transcription. J. Virol. 1993, 67, 1916–1926. [Google Scholar] [PubMed]
- Lemm, J.A.; Rice, C.M. Assembly of functional Sindbis virus RNA replication complexes: Requirement for coexpression of P123 and P34. J. Virol. 1993, 67, 1905–1915. [Google Scholar] [PubMed]
- Lemm, J.A.; Rumenapf, T.; Strauss, E.G.; Strauss, J.H.; Rice, C.M. Polypeptide requirements for assembly of functional Sindbis virus replication complexes: A model for the temporal regulation of minus- and plus-strand RNA synthesis. EMBO J. 1994, 13, 2925–2934. [Google Scholar] [PubMed]
- Ahola, T.; Kääriäinen, L. Reaction in alphavirus mRNA capping: Formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc. Natl. Acad. Sci. USA 1995, 92, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Spuul, P.; Salonen, A.; Merits, A.; Jokitalo, E.; Kaariainen, L.; Ahola, T. Role of the amphipathic peptide of Semliki forest virus replicase protein nsP1 in membrane association and virus replication. J. Virol. 2007, 81, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Das, P.K.; Merits, A.; Lulla, A. Functional cross-talk between distant domains of chikungunya virus non-structural protein 2 is decisive for its RNA-modulating activity. J. Biol. Chem. 2014, 289, 5635–5653. [Google Scholar] [CrossRef] [PubMed]
- Hardy, W.R.; Strauss, J.H. Processing the nonstructural polyproteins of sindbis virus: Nonstructural proteinase is in the C-terminal half of nsP2 and functions both in cis and in trans. J. Virol. 1989, 63, 4653–4664. [Google Scholar] [PubMed]
- Vasiljeva, L.; Merits, A.; Golubtsov, A.; Sizemskaja, V.; Kaariainen, L.; Ahola, T. Regulation of the sequential processing of Semliki Forest virus replicase polyprotein. J. Biol. Chem. 2003, 278, 41636–41645. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Reynaud, J.M.; Rasalouskaya, A.; Akhrymuk, I.; Mobley, J.A.; Frolov, I.; Frolova, E.I. New world and old world alphaviruses have evolved to exploit different components of stress granules, FXR and G3BP proteins, for assembly of viral replication complexes. PLoS Pathog. 2016, 12, e1005810. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Debing, Y.; Jankevicius, G.; Neyts, J.; Ahel, I.; Coutard, B.; Canard, B. Viral macro domains reverse protein ADP-ribosylation. J. Virol. 2016, 90, 8478–8486. [Google Scholar] [CrossRef] [PubMed]
- Rubach, J.K.; Wasik, B.R.; Rupp, J.C.; Kuhn, R.J.; Hardy, R.W.; Smith, J.L. Characterization of purified Sindbis virus nsP4 RNA-dependent RNA polymerase activity in vitro. Virology 2009, 384, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Bartenschlager, R. Architecture and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2013, 2, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Spuul, P.; Balistreri, G.; Kääriäinen, L.; Ahola, T. Phosphatidylinositol 3-kinase-, actin-, and microtubule-dependent transport of Semliki Forest virus replication complexes from the plasma membrane to modified lysosomes. J. Virol. 2010, 84, 7543–7557. [Google Scholar] [CrossRef] [PubMed]
- Frolova, E.I.; Gorchakov, R.; Pereboeva, L.; Atasheva, S.; Frolov, I. Functional Sindbis virus replicative complexes are formed at the plasma membrane. J. Virol. 2010, 84, 11679–11695. [Google Scholar] [CrossRef] [PubMed]
- Thaa, B.; Biasiotto, R.; Eng, K.; Neuvonen, M.; Gotte, B.; Rheinemann, L.; Mutso, M.; Utt, A.; Varghese, F.; Balistreri, G.; et al. Differential phosphatidylinositol-3-kinase-Akt-mTOR activation by Semliki Forest and Chikungunya viruses is dependent on nsP3 and connected to replication complex internalization. J. Virol. 2015, 89, 11420–11437. [Google Scholar] [CrossRef] [PubMed]
- Shirako, Y.; Strauss, J.H. Regulation of Sindbis virus RNA replication: Uncleaved P123 and nsP4 function in minus-strand RNA synthesis, whereas cleaved products from P123 are required for efficient plus-strand RNA synthesis. J. Virol. 1994, 68, 1874–1885. [Google Scholar] [PubMed]
- Kallio, K.; Hellström, K.; Balistreri, G.; Spuul, P.; Jokitalo, E.; Ahola, T. Template RNA length determines the size of replication complex spherules for Semliki Forest virus. J. Virol. 2013, 87, 9125–9134. [Google Scholar] [CrossRef] [PubMed]
- Ertel, K.J.; Benefield, D.; Castano-Diez, D.; Pennington, J.G.; Horswill, M.; den Boon, J.A.; Otegui, M.S.; Ahlquist, P. Cryo-electron tomography reveals novel features of a viral RNA replication compartment. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Kallio, K.; Hellström, K.; Jokitalo, E.; Ahola, T. RNA replication and membrane modification require the same functions of alphavirus nonstructural proteins. J. Virol. 2015, 90, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Albulescu, I.C.; Tas, A.; Scholte, F.E.; Snijder, E.J.; van Hemert, M.J. An in vitro assay to study chikungunya virus RNA synthesis and the mode of action of inhibitors. J. Gen. Virol. 2014, 95, 2683–2692. [Google Scholar] [CrossRef] [PubMed]
- Spuul, P.; Balistreri, G.; Hellström, K.; Golubtsov, A.V.; Jokitalo, E.; Ahola, T. Assembly of alphavirus replication complexes from RNA and protein components in a novel trans-replication system in mammalian cells. J. Virol. 2011, 85, 4739–4751. [Google Scholar] [CrossRef] [PubMed]
- Hellström, K.; Kallio, K.; Meriläinen, H.M.; Jokitalo, E.; Ahola, T. Ability of minus strands and modified plus strands to act as templates in Semliki Forest virus RNA replication. J. Gen. Virol. 2016, 97, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Utt, A.; Quirin, T.; Saul, S.; Hellström, K.; Ahola, T.; Merits, A. Versatile trans-replication systems for Chikungunya virus allow functional analysis and tagging of every replicase protein. PLoS ONE 2016, 11, e0151616. [Google Scholar] [CrossRef] [PubMed]
- Hellström, K.; Kallio, K.; Utt, A.; Quirin, T.; Jokitalo, E.; Merits, A.; Ahola, T. Partially uncleaved alphavirus replicase forms spherule structures in the presence and absence of RNA template. J. Virol. 2017, 91, e00787. [Google Scholar] [CrossRef] [PubMed]
- Lemm, J.A.; Bergqvist, A.; Read, C.M.; Rice, C.M. Template-dependent initiation of Sindbis virus RNA replication in vitro. J. Virol. 1998, 72, 6546–6553. [Google Scholar] [PubMed]
- Barton, D.J.; Sawicki, S.G.; Sawicki, D.L. Solubilization and immunoprecipitation of alphavirus replication complexes. J. Virol. 1991, 65, 1496–1506. [Google Scholar] [PubMed]
- Clewley, J.P.; Kennedy, S.I. Purification and polypeptide composition of Semliki Forest virus RNA polymerase. J. Gen. Virol. 1976, 32, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Wielgosz, M.M.; Huang, H.V. A novel viral RNA species in Sindbis virus-infected cells. J. Virol. 1997, 71, 9108–9117. [Google Scholar] [PubMed]
- Buchholz, U.J.; Finke, S.; Conzelmann, K.K. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 1999, 73, 251–259. [Google Scholar] [PubMed]
- Ulper, L.; Sarand, I.; Rausalu, K.; Merits, A. Construction, properties, and potential application of infectious plasmids containing Semliki Forest virus full-length cDNA with an inserted intron. J. Virol. Methods 2008, 148, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Keränen, S.; Kääriäinen, L. Isolation and basic characterization of temperature-sensitive mutants from Semliki Forest virus. Acta. Pathol. Microbiol. Scand. B Microbiol. Immunol. 1974, 82, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Scholte, F.E.; Tas, A.; Martina, B.E.; Cordioli, P.; Narayanan, K.; Makino, S.; Snijder, E.J.; van Hemert, M.J. Characterization of synthetic Chikungunya viruses based on the consensus sequence of recent E1–226V isolates. PLoS ONE 2013, 8, e71047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, D.; Hoppe, S.; Saher, G.; Krijnse-Locker, J.; Bartenschlager, R. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 2013, 87, 10612–10627. [Google Scholar] [CrossRef] [PubMed]
- Van Dinten, L.C.; den Boon, J.A.; Wassenaar, A.L.; Spaan, W.J.; Snijder, E.J. An infectious arterivirus cDNA clone: Identification of a replicase point mutation that abolishes discontinuous mRNA transcription. Proc. Natl. Acad. Sci. USA 1997, 94, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Kujala, P.; Ikäheimonen, A.; Ehsani, N.; Vihinen, H.; Auvinen, P.; Kääriäinen, L. Biogenesis of the Semliki Forest virus RNA replication complex. J. Virol. 2001, 75, 3873–3884. [Google Scholar] [CrossRef] [PubMed]
- Van Hemert, M.J.; van den Worm, S.H.; Knoops, K.; Mommaas, A.M.; Gorbalenya, A.E.; Snijder, E.J. SARS-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathog. 2008, 4, e1000054. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, D.L.; Sawicki, S.G. Short-lived minus-strand polymerase for Semliki Forest virus. J. Virol. 1980, 34, 108–118. [Google Scholar] [PubMed]
- Sawicki, S.G.; Sawicki, D.L. The effect of overproduction of nonstructural proteins on alphavirus plus-strand and minus-strand RNA synthesis. Virology 1986, 152, 507–512. [Google Scholar] [CrossRef]
- Gorchakov, R.; Frolova, E.; Sawicki, S.; Atasheva, S.; Sawicki, D.; Frolov, I. A new role for ns polyprotein cleavage in Sindbis virus replication. J. Virol. 2008, 82, 6218–6231. [Google Scholar] [CrossRef] [PubMed]
- Frolova, E.; Gorchakov, R.; Garmashova, N.; Atasheva, S.; Vergara, L.A.; Frolov, I. Formation of nsP3-specific protein complexes during Sindbis virus replication. J. Virol. 2006, 80, 4122–4134. [Google Scholar] [CrossRef] [PubMed]
- Gorchakov, R.; Garmashova, N.; Frolova, E.; Frolov, I. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells. J. Virol. 2008, 82, 10088–10101. [Google Scholar] [CrossRef] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietilä, M.K.; Albulescu, I.C.; Hemert, M.J.v.; Ahola, T. Polyprotein Processing as a Determinant for in Vitro Activity of Semliki Forest Virus Replicase. Viruses 2017, 9, 292. https://doi.org/10.3390/v9100292
Pietilä MK, Albulescu IC, Hemert MJv, Ahola T. Polyprotein Processing as a Determinant for in Vitro Activity of Semliki Forest Virus Replicase. Viruses. 2017; 9(10):292. https://doi.org/10.3390/v9100292
Chicago/Turabian StylePietilä, Maija K., Irina C. Albulescu, Martijn J. van Hemert, and Tero Ahola. 2017. "Polyprotein Processing as a Determinant for in Vitro Activity of Semliki Forest Virus Replicase" Viruses 9, no. 10: 292. https://doi.org/10.3390/v9100292
APA StylePietilä, M. K., Albulescu, I. C., Hemert, M. J. v., & Ahola, T. (2017). Polyprotein Processing as a Determinant for in Vitro Activity of Semliki Forest Virus Replicase. Viruses, 9(10), 292. https://doi.org/10.3390/v9100292