Innate Immune Evasion Mediated by Flaviviridae Non-Structural Proteins


Abstract
:1. Introduction
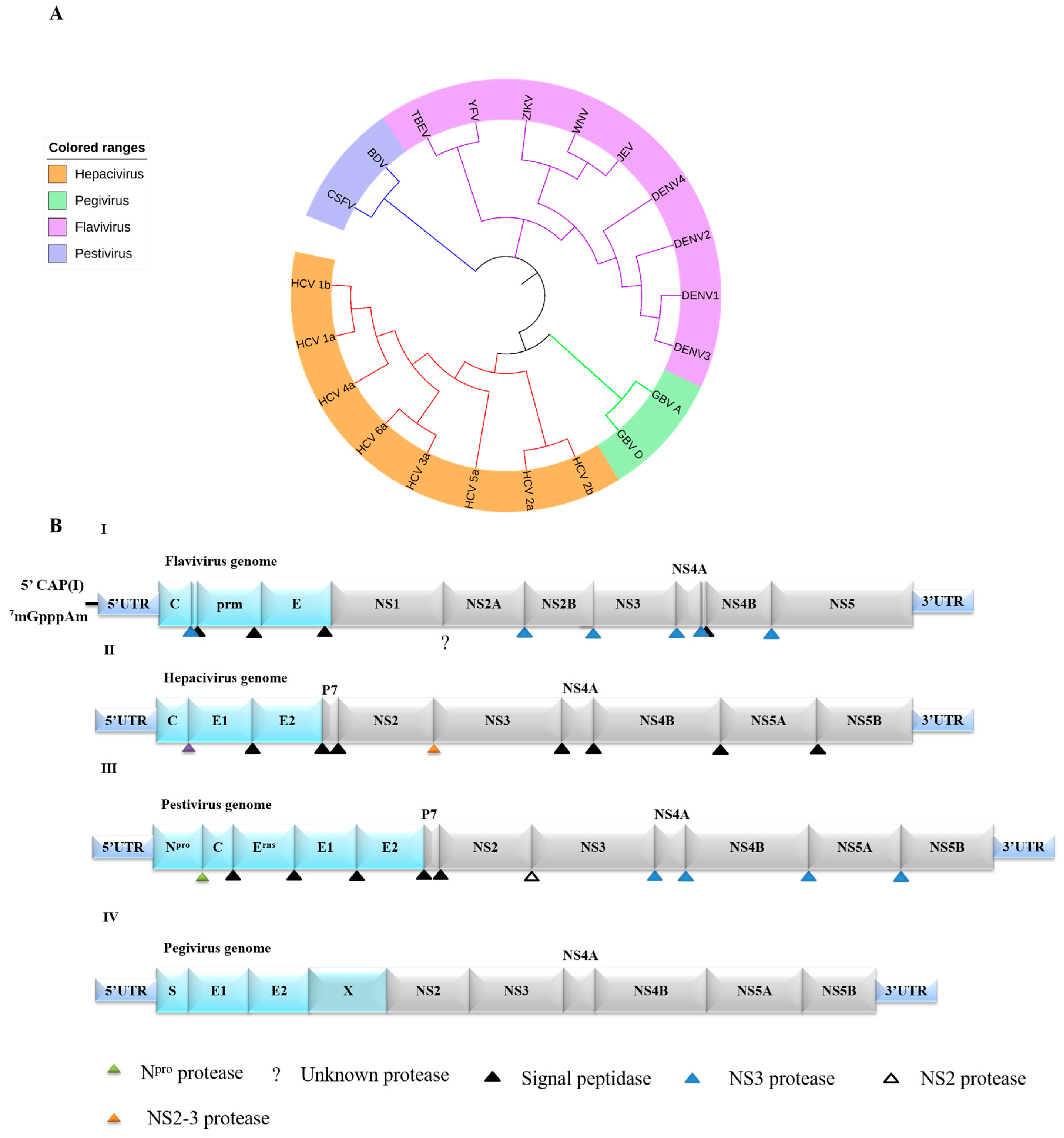
2. Genome and Life Cycle of Flaviviridae
3. Innate Sensing of Viruses by Pattern Recognition Receptors
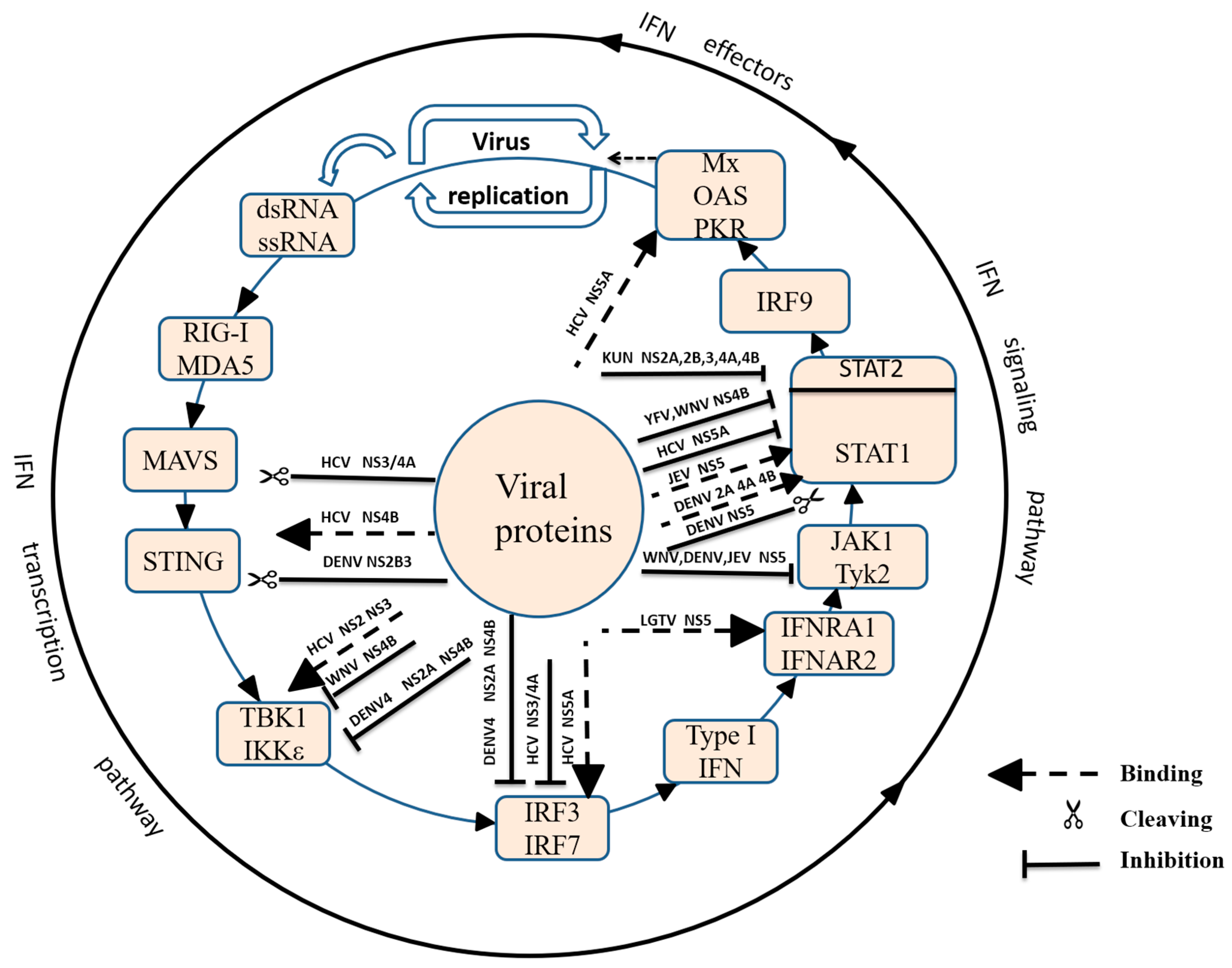
4. Flaviviridae Antagonism, IFN Induction and IFN-Dependent Signalling Pathways
4.1. Non-Structural Protein 1 (NS1) Inhibits the TLR Signalling Pathway
4.2. Non-Structural Protein 2 (NS2)
4.2.1. Suppression of IFN-β Induction
4.2.2. Suppression of the IFN-Dependent Signalling Pathway
4.3. Non-Structural Protein 3 (NS3)
4.3.1. HCV NS3 Evades Innate Immunity
4.3.2. The Role of Protease Complex NS2B3 and NS3/4A in Inhibiting Innate Immunity
4.4. Non-Structural Proteins 4A and 4B (NS4A and NS4B)
4.4.1. HCV NS4B Inhibits IFN-β Induction by Interacting with STING
4.4.2. NS4B Inhibits the IFN-Dependent Signalling Pathway
4.5. Non-Structural Protein 5 (NS5)
4.5.1. HCV NS5A Inhibits IFNβ Induction
4.5.2. Flaviviridae NS5 Inhibits the IFN-Dependent Signalling Pathway
5. Conclusions
Acknowledgements
Conflicts of Interest
References
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef] [PubMed]
- Kane, A.; Lloyd, J.; Zaffran, M.; Simonsen, L.; Kane, M. Transmission of hepatitis B, hepatitis C and human immunodeficiency viruses through unsafe injections in the developing world: Model-based regional estimates. Bull. World Health Organ. 1969, 77, 801–807. [Google Scholar]
- Gaunt, M.W.; Sall, A.A.; de Lamballerie, X.; Falconar, A.K.; Dzhivanian, T.I.; Gould, E.A. Phylogenetic relationships of flaviviruses correlate with their epidemiology, disease association and biogeography. J. Gen. Virol. 2001, 82, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- De, F.M.; Ulbert, S.; Diamond, M.; Sanders, N.N. Recent progress in West Nile virus diagnosis and vaccination. Vet. Res. 2012, 43, 16. [Google Scholar] [Green Version]
- Bressanelli, S.; Stiasny, K.; Allison, S.L.; Stura, E.A.; Duquerroy, S.; Lescar, J.; Heinz, F.X.; Rey, F.A. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 2004, 23, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Rice, C.M. Flaviviridae: The viruses and their replication. Fields Virology 1996, 3, 931–959. [Google Scholar]
- Heinz, F.X.; Auer, G.; Stiasny, K.; Holzmann, H.; Mandl, C.; Guirakhoo, F.; Kunz, C. The interactions of the flavivirus envelope proteins: Implications for virus entry and release. Arch. Virol. Suppl. 1994, 9, 339–348. [Google Scholar] [PubMed]
- Heinz, F.X.; Stiasny, K.; Püschnerauer, G.; Holzmann, H.; Allison, S.L.; Mandl, C.W.; Kunz, C. Structural changes and functional control of the tick-borne encephalitis virus glycoprotein E by the heterodimeric association with protein prM. Virology 1994, 198, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Khromykh, A.A.; Sedlak, P.L.; Westaway, E.G. trans-Complementation analysis of the flavivirus Kunjin ns5 gene reveals an essential role for translation of its N-terminal half in RNA replication. J. Virol. 1999, 73, 9247–9255. [Google Scholar] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Barton, G.M. Viral recognition by Toll-like receptors. Semin Immunol. 2007, 19, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Green, A.M.; Beatty, P.R.; Hadjilaou, A.; Harris, E. Innate immunity to dengue virus infection and subversion of antiviral responses. J. Mol. Biol. 2014, 426, 1148–1160. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.T.; Mountain, A.J.; Kelly, M.P.; Hall, C.; Rigopoulos, A.; Johns, T.G.; Smyth, F.E.; Brechbiel, M.W.; Nice, E.C.; Burgess, A.W.; et al. Enhanced efficacy of radioimmunotherapy with 90Y-CHX-A''-DTPA-hu3S193 by inhibition of epidermal growth factor receptor (EGFR) signaling with EGFR tyrosine kinase inhibitor AG1478. Clin. Cancer Res. 2005, 11, S7080–S7086. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and MDA5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Tu, D.; Zhu, Z.; Zhou, A.Y.; Yun, C.H.; Lee, K.E.; Toms, A.V.; Li, Y.; Dunn, G.P.; Chan, E.; Thai, T.; et al. Structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep. 2013, 3, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Tasaka, M.; Sakamoto, N.; Itakura, Y.; Nakagawa, M.; Itsui, Y.; Sekine-Osajima, Y.; Nishimura-Sakurai, Y.; Chen, C.H.; Yoneyama, M.; Fujita, T.; et al. Hepatitis C virus non-structural proteins responsible for suppression of the RIG-I/Cardif-induced interferon response. J. Gen. Virol. 2007, 88, 3323–3333. [Google Scholar] [CrossRef] [PubMed]
- Hiscott, J. Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 2007, 282, 15325–15329. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, J.L.; Baltimore, D. NF-κB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999, 18, 6694–6704. [Google Scholar] [CrossRef] [PubMed]
- Paz, S.; Sun, Q.; Nakhaei, P.; Romieu-Mourez, R.; Goubau, D.; Julkunen, I.; Lin, R.; Hiscott, J. Induction of IRF-3 and IRF-7 phosphorylation following activation of the RIG-I pathway. Cell Mol. Biol. (Noisy-le-grand) 2006, 52, 17–28. [Google Scholar]
- Gleason, C.E.; Ordureau, A.; Gourlay, R.; Arthur, J.S.; Cohen, P. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon β. J. Biol. Chem. 2011, 286, 35663–35674. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Helgason, E.; Phung, Q.T.; Quan, C.L.; Iyer, R.S.; Lee, M.W.; Bowman, K.K.; Starovasnik, M.A.; Dueber, E.C. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc. Natl. Acad. Sci. USA 2012, 109, 9378–9383. [Google Scholar] [CrossRef] [PubMed]
- Van Boxel-Dezaire, A.H.; Rani, M.R.; Stark, G.R. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 2006, 25, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Chen, Z.J. MITAgating viral infection. Immunity 2008, 29, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Nakhaei, P.; Hiscott, J.; Lin, R. STING-ing the antiviral pathway. J. Mol. Cell Biol. 2010, 2, 110–112. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Kato, H.; Sato, S.; Takahashi, K.; Coban, C.; Yamamoto, M.; Uematsu, S.; Ishii, K.J.; Takeuchi, O.; et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 2006, 203, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Roberts, T.G.; Edgil, D.; Lu, B.; Ernst, J.; Harris, E. Modulation of Dengue virus infection in human cells by α, β, and γ interferons. J. Virol. 2000, 74, 4957–4966. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.H.; Moradpour, D.; Blum, H.E. Expression of Hepatitis C Virus Proteins Inhibits Signal Transduction through the Jak-STAT Pathway. J. Virol. 1999, 73, 8469–8475. [Google Scholar] [PubMed]
- Chambers, T.J.; McCourt, D.W.; Rice, C.M. Production of yellow fever virus proteins in infected cells: identification of discrete polyprotein species and analysis of cleavage kinetics using region-specific polyclonal antisera. Virology 1990, 177, 159–174. [Google Scholar] [CrossRef]
- Muylaert, I.R.; Chambers, T.J.; Galler, R.; Rice, C.M. Mutagenesis of the N-linked glycosylation sites of the yellow fever virus NS1 protein: Effects on virus replication and mouse neurovirulence. Virology 1996, 222, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Liu, Y.; Yuan, Z. Critical role of Dengue Virus NS1 protein in viral replication. Virol. Sin. 2014, 29, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Blitvich, B.; Mackenzie, J.; Coelen, R.; Howard, M.; Hall, R. A novel complex formed between the flavivirus E and NS1 proteins: Analysis of its structure and function. Arch. Virol. 1995, 140, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Lindenbach, B.D.; Pragai, B.M.; McCourt, D.W.; Rice, C.M. Processing in the hepatitis C virus E2-NS2 region: Identification of p7 and two distinct E2-specific products with different C termini. J. Virol. 1994, 68, 5063–5073. [Google Scholar] [PubMed]
- Elbers, K.; Tautz, N.; Becher, P.; Stoll, D.; Rumenapf, T.; Thiel, H.J. Processing in the pestivirus E2-NS2 region: Identification of proteins p7 and E2p7. J. Virol. 1996, 70, 4131–4135. [Google Scholar] [PubMed]
- Schlesinger, J.J.; Foltzer, M.; Chapman, S. The Fc portion of antibody to yellow fever virus NS1 is a determinant of protection against YF encephalitis in mice. Virology 1993, 192, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.A.; Buckley, A.; Barrett, A.D.; Cammack, N. Neutralizing (54K) and non-neutralizing (54K and 48K) monoclonal antibodies against structural and non-structural yellow fever virus proteins confer immunity in mice. J. Gen. Virol. 1986, 67, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Modhiran, N.; Watterson, D.; Muller, D.A.; Panetta, A.K.; Sester, D.P.; Liu, L.; Hume, D.A.; Stacey, K.J.; Young, P.R. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Beatty, P.R.; Puerta-Guardo, H.; Killingbeck, S.S.; Glasner, D.R.; Hopkins, K.; Harris, E. Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.R.; de Sessions, P.F.; Leon, M.A.; Scholle, F. West Nile virus nonstructural protein 1 inhibits TLR3 signal transduction. J. Virol. 2008, 82, 8262–8271. [Google Scholar] [CrossRef] [PubMed]
- Fredericksen, B.L.; Jr, G.M. West Nile virus evades activation of interferon regulatory factor 3 through RIG-I-dependent and -independent pathways without antagonizing host defense signaling. J. Virol. 2006, 80, 2913–2923. [Google Scholar] [CrossRef] [PubMed]
- Baronti, C.; Sire, J.; de Lamballerie, X.; Querat, G. Nonstructural NS1 proteins of several mosquito-borne Flavivirus do not inhibit TLR3 signaling. Virology 2010, 404, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Santolini, E.; Pacini, L.; Fipaldini, C.; Migliaccio, G.; Monica, N. The NS2 protein of hepatitis C virus is a transmembrane polypeptide. J. Virol. 1995, 69, 7461–7471. [Google Scholar] [PubMed]
- De Moerlooze, L.; Desport, M.; Renard, A.; Lecomte, C.; Brownlie, J.; Martial, J.A. The coding region for the 54-kDa protein of several pestiviruses lacks host insertions but reveals a “zinc finger-like” domain. Virology 1990, 177, 812–815. [Google Scholar] [CrossRef]
- Erdtmann, L.; Franck, N.; Lerat, H.; Le Seyec, J.; Gilot, D.; Cannie, I.; Gripon, P.; Hibner, U.; Guguen-Guillouzo, C. The hepatitis C virus NS2 protein is an inhibitor of CIDE-B-induced apoptosis. J. Biol. Chem. 2003, 278, 18256–18264. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Liu, J.; Ye, L.; Liao, Q.J.; Wu, J.G.; Gao, J.R.; She, Y.L.; Wu, Z.H.; Ye, L.B. HCV NS2 protein inhibits cell proliferation and induces cell cycle arrest in the S-phase in mammalian cells through down-regulation of cyclin A expression. Virus Res. 2006, 121, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Chen, H.B.; Wang, X.J.; Huang, H.; Khromykh, A.A. Analysis of adaptive mutations in Kunjin virus replicon RNA reveals a novel role for the flavivirus nonstructural protein NS2A in inhibition of β interferon promoter-driven transcription. J. Virol. 2004, 78, 12225–12235. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Jordan, J.L.; Sanchez-Burgos, G.G.; Laurent-Rolle, M.; Garcia-Sastre, A. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA 2003, 100, 14333–14338. [Google Scholar] [CrossRef] [PubMed]
- Dalrymple, N.A.; Cimica, V.; Mackow, E.R. Dengue Virus NS Proteins Inhibit RIG-I/MAVS Signaling by Blocking TBK1/IRF3 Phosphorylation: Dengue Virus Serotype 1 NS4A Is a Unique Interferon-Regulating Virulence Determinant. MBio. 2015, 6, e00553-15. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, S.; Maestre, A.M.; Pagni, S.; Patel, J.R.; Savage, T.; Gutman, D.; Maringer, K.; Bernalrubio, D.; Shabman, R.S.; Simon, V. DENV Inhibits Type I IFN Production in Infected Cells by Cleaving Human STING. PLoS Pathog. 2011, 8, e1002934. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.Y.; Chang, T.H.; Liang, J.J.; Chiang, R.L.; Lee, Y.L.; Liao, C.L.; Lin, Y.L. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog. 2012, 8, e1002780. [Google Scholar] [CrossRef] [PubMed]
- Kaukinen, P.; Sillanpaa, M.; Nousiainen, L.; Melen, K.; Julkunen, I. Hepatitis C virus NS2 protease inhibits host cell antiviral response by inhibiting IKKepsilon and TBK1 functions. J. Med. Virol. 2013, 85, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Wang, X.J.; Mokhonov, V.V.; Shi, P.Y.; Randall, R.; Khromykh, A.A. Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins. J. Virol. 2005, 79, 1934. [Google Scholar] [CrossRef] [PubMed]
- Steinkühler, C.; Tomei, L.; de Francesco, R. In vitro activity of hepatitis C virus protease NS3 purified from recombinant Baculovirus-infected Sf9 cells. J. Biol. Chem. 1996, 271, 6367–6373. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Sato, M.; Chieda, S.; Shoji, I.; Harada, T.; Yamakawa, Y.; Watabe, S.; Matsuura, Y.; Miyamura, T. In vivo and in vitro trans-cleavage activity of hepatitis C virus serine proteinase expressed by recombinant baculoviruses. J. Gen. Virol. 1995, 76, 3021–3029. [Google Scholar] [CrossRef] [PubMed]
- Bouffard, P.; Bartenschlager, R.; Ahlborn-Laake, L.; Mous, J.; Roberts, N.; Jacobsen, H. An in vitro assay for hepatitis C virus NS3 serine proteinase. Virology 1995, 209, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Preugschat, F.; Averett, D.R.; Clarke, B.E.; Porter, D.J. A steady-state and pre-steady-state kinetic analysis of the NTPase activity associated with the hepatitis C virus NS3 helicase domain. J. Biol. Chem. 1996, 271, 24449–24457. [Google Scholar] [CrossRef] [PubMed]
- Suzich, J.A.; Tamura, J.K.; Palmer-Hill, F.; Warrener, P.; Grakoui, A.; Rice, C.M.; Feinstone, S.M.; Collett, M.S. Hepatitis C virus NS3 protein polynucleotide-stimulated nucleoside triphosphatase and comparison with the related pestivirus and flavivirus enzymes. J. Virol. 1993, 67, 6152–6158. [Google Scholar] [PubMed]
- Warrener, P.; Collett, M.S. Pestivirus NS3 (p80) protein possesses RNA helicase activity. J. Virol. 1995, 69, 1720–1726. [Google Scholar] [PubMed]
- Ling, J.; Peterson, D.L. Expression, Isolation, and Characterization of the Hepatitis C Virus ATPase/RNA Helicase. Arch. Biochem. Biophys. 1995, 323, 47–53. [Google Scholar]
- Tai, C.L.; Chi, W.K.; Chen, D.S.; Hwang, L.H. The helicase activity associated with hepatitis C virus nonstructural protein 3 (NS3). J. Virol. 1996, 70, 8477–8484. [Google Scholar] [PubMed]
- Gwack, Y.; Dong, W.K.; Han, J.H.; Choe, J. Characterization of RNA Binding Activity and RNA Helicase Activity of the Hepatitis C Virus NS3 Protein. Biochem. Biophys. Res. Commun. 1996, 225, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Cahour, A.; Falgout, B.; Lai, C.J. Cleavage of the dengue virus polyprotein at the NS3/NS4A and NS4B/NS5 junctions is mediated by viral protease NS2B-NS3, whereas NS4A/NS4B may be processed by a cellular protease. J. Virol. 1992, 66, 1535–1542. [Google Scholar] [PubMed]
- Tanji, Y.; Hijikata, M.; Satoh, S.; Kaneko, T.; Shimotohno, K. Hepatitis C virus-encoded nonstructural protein NS4A has versatile functions in viral protein processing. J. Virol. 1995, 69, 1575–1581. [Google Scholar] [PubMed]
- Butkiewicz, N.J.; Wendel, M.; Zhang, R.; Jubin, R.; Pichardo, J.; Smith, E.B.; Hart, A.M.; Ingram, R.; Durkin, J.; Mui, P.W.; et al. Enhancement of hepatitis C virus NS3 proteinase activity by association with NS4A-specific synthetic peptides: Identification of sequence and critical residues of NS4A for the cofactor activity. Virology 1996, 225, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Satoh, S.; Tanji, Y.; Hijikata, M.; Kimura, K.; Shimotohno, K. The N-terminal region of hepatitis C virus nonstructural protein 3 (NS3) is essential for stable complex formation with NS4A. J. Virol. 1995, 69, 4255–4260. [Google Scholar] [PubMed]
- Shimizu, Y.; Yamaji, K.; Masuho, Y.; Yokota, T.; Inoue, H.; Sudo, K.; Satoh, S.; Shimotohno, K. Identification of the sequence on NS4A required for enhanced cleavage of the NS5A/5B site by hepatitis C virus NS3 protease. J. Virol. 1996, 70, 127–132. [Google Scholar] [PubMed]
- Tomei, L.; Failla, C.; Vitale, R.L.; Bianchi, E.; de Francesco, R. A central hydrophobic domain of the hepatitis C virus NS4A protein is necessary and sufficient for the activation of the NS3 protease. J. Gen. Virol. 1996, 77, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Yamshchikov, V.F.; Compans, R.W. Formation of the flavivirus envelope: Role of the viral NS2B-NS3 protease. J. Virol. 1995, 69, 1995–2003. [Google Scholar] [PubMed]
- Jan, L.R.; Yang, C.S.; Trent, D.W.; Falgout, B.; Lai, C.J. Processing of Japanese encephalitis virus non-structural proteins: NS2B-NS3 complex and heterologous proteases. J. Gen. Virol. 1995, 76, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Kato, N.; Moriyama, M.; Taniguchi, H.; Wang, Y.; Dharel, N.; Kawabe, T.; Omata, M. Interaction between the HCV NS3 protein and the host TBK1 protein leads to inhibition of cellular antiviral responses. Hepatology 2005, 41, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat. Cell Biol. 2009, 11, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; He, L.; Peng, Y.; Shi, X.; Chen, J.; Zhong, J.; Chen, X.; Cheng, G.; Deng, H. The hepatitis C virus protein NS3 suppresses TNF-α-stimulated activation of NF-κB by targeting LUBAC. Sci. Signal 2015, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed]
- Foy, E.; Li, K.; Wang, C.; Jr, S.R.; Ikeda, M.; Lemon, S.M.; Jr, G.M. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 2003, 300, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M.; Chambers, T.J.; Monath, T.P. Molecular biology of flaviviruses. Adv. Virus Res. 2003, 59, 23–61. [Google Scholar] [PubMed]
- Nakaya, T.; Cros, J.; Park, M.S.; Nakaya, Y.; Zheng, H.; Sagrera, A.; Villar, E.; García-Sastre, A.; Palese, P. Recombinant Newcastle disease virus as a vaccine vector. J. Virol. 2001, 75, 11868–11873. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Madoz, J.R.; Belicha-Villanueva, A.; Bernal-Rubio, D.; Ashour, J.; Ayllon, J.; Fernandez-Sesma, A. Inhibition of the type I interferon response in human dendritic cells by dengue virus infection requires a catalytically active NS2B3 complex. J. Virol. 2010, 84, 9760–9774. [Google Scholar] [CrossRef] [PubMed]
- Yusof, R.; Clum, S.; Wetzel, M.; Murthy, H.M.; Padmanabhan, R. Purified NS2B/NS3 serine protease of dengue virus type 2 exhibits cofactor NS2B dependence for cleavage of substrates with dibasic amino acids in vitro. J. Biol. Chem. 2000, 275, 9963–9969. [Google Scholar] [CrossRef] [PubMed]
- Khumthong, R.; Angsuthanasombat, C.; Panyim, S.; Katzenmeier, G. In vitro determination of dengue virus type 2 NS2B-NS3 protease activity with fluorescent peptide substrates. J. Biochem. Mol. Biol. 2002, 35, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Jordan, J.L.; Laurent-Rolle, M.; Ashour, J.; Martinez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; Garcia-Sastre, A. Inhibition of α/β interferon signaling by the NS4B protein of flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar] [CrossRef] [PubMed]
- Nitta, S.; Sakamoto, N.; Nakagawa, M.; Kakinuma, S.; Mishima, K.; Kusano-Kitazume, A.; Kiyohashi, K.; Murakawa, M.; Nishimura-Sakurai, Y.; Azuma, S.; et al. Hepatitis C virus NS4B protein targets STING and abrogates RIG-I-mediated type I interferon-dependent innate immunity. Hepatology 2013, 57, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Cao, X.; Lu, J.; Huang, B.; Liu, Y.J.; Kato, N.; Shu, H.B.; Zhong, J. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J. Hepatol. 2013, 59, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Yi, G.; Wen, Y.; Shu, C.; Han, Q.; Konan, K.V.; Li, P.; Kao, C.C. Hepatitis C Virus NS4B Can Suppress STING Accumulation To Evade Innate Immune Responses. J. Virol. 2016, 90, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Kumthip, K.; Chusri, P.; Jilg, N.; Zhao, L.; Fusco, D.N.; Zhao, H.; Goto, K.; Cheng, D.; Schaefer, E.A.; Zhang, L.; et al. Hepatitis C virus NS5A disrupts STAT1 phosphorylation and suppresses type I interferon signaling. J. Virol. 2012, 86, 8581–8591. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Chen, Z.J. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal 2012, 5, ra20. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Kuo, M.D.; Chien, L.J.; Hsu, S.L.; Wang, Y.M.; Lin, J.H. RNA-protein interactions: Involvement of NS3, NS5, and 3´ noncoding regions of Japanese encephalitis virus genomic RNA. J. Virol. 1997, 71, 3466–3473. [Google Scholar] [PubMed]
- Tan, B.H.; Jianlin, F.U.; Sugrue, R.J.; Yap, E.H.; Chan, Y.C.; Tan, Y.H. Recombinant Dengue Type 1 Virus NS5 Protein Expressed in Escherichia coli Exhibits RNA-Dependent RNA Polymerase Activity. Virology 1996, 216, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Behrens, S.E.; Tomei, L.; de Francesco, R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 1996, 15, 12–22. [Google Scholar] [PubMed]
- Hwang, S.B.; Park, K.J.; Kim, Y.S.; Sung, Y.C.; Lai, M.M. Hepatitis C Virus NS5B Protein Is a Membrane-Associated Phosphoprotein with a Predominantly Perinuclear Localization. Virology 1997, 227, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.H.; Kumar, U.; Thomas, H.C.; Wen, Y.M.; Monjardino, J. Expression, purification, and partial characterization of HCV RNA polymerase. Biochem. Biophys. Res. Commun. 1997, 232, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Egloff, M.P.; Benarroch, D.; Selisko, B.; Romette, J.L.; Canard, B. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: Crystal structure and functional characterization. EMBO J. 2002, 21, 2757–2768. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Shah, A.; Tilgner, M.; Guo, Y.; Zhao, Y.; Dong, H.; Deas, T.S.; Zhou, Y.; Li, H.; Shi, P.Y. West Nile virus 5′-cap structure is formed by sequential guanine N-7 and ribose 2′-O methylations by nonstructural protein 5. J. Virol. 2006, 80, 8362–8370. [Google Scholar] [CrossRef] [PubMed]
- Kamer, G.; Argos, P. Primary structural comparison of RNA-dependent polymerases from plant, animal and bacterial viruses. Nucleic Acids Res. 2011, 12, 7269–7282. [Google Scholar] [CrossRef]
- Miller, R.H.; Purcell, R.H. Hepatitis C virus shares amino acid sequence similarity with pestiviruses and flaviviruses as well as members of two plant virus supergroups. Proc. Natl. Acad. Sci. USA 1990, 87, 2057–2061. [Google Scholar] [CrossRef] [PubMed]
- Ide, Y.; Zhang, L.; Chen, M.; Inchauspe, G.; Bahl, C.; Sasaguri, Y.; Padmanabhan, R. Characterization of the nuclear localization signal and subcellular distribution of hepatitis C virus nonstructural protein NS5A. Gene 1996, 182, 203–211. [Google Scholar] [CrossRef]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.Y.; García-Sastre, A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, J.B.; Kim, H.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A protein modulates IRF-7-mediated interferon-α signaling. J. Interferon Cytokine Res. 2014, 34, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Best, S.M.; Morris, K.L.; Shannon, J.G.; Robertson, S.J.; Mitzel, D.N.; Park, G.S.; Boer, E.; Wolfinbarger, J.B.; Bloom, M.E. Inhibition of Interferon-Stimulated JAK-STAT Signaling by a Tick-Borne Flavivirus and Identification of NS5 as an Interferon Antagonist. J. Virol. 2005, 79, 12828–12839. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Rolle, M.; Morrison, J.; Rajsbaum, R.; Macleod, J.M.; Pisanelli, G.; Pham, A.; Ayllon, J.; Miorin, L.; Martínez-Romero, C.; Tenoever, B.R.; et al. The interferon signaling antagonist function of yellow fever virus NS5 protein is activated by type I interferon. Cell Host Microbe 2014, 16, 314–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sanchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Kaname, Y.; Hamamoto, I.; Tsuda, Y.; Wen, X.; Taguwa, S.; Moriishi, K.; Takeuchi, O.; Kawai, T.; Kanto, T.; et al. Hepatitis C virus nonstructural protein 5A modulates the toll-like receptor-MyD88-dependent signaling pathway in macrophage cell lines. J. Virol. 2007, 81, 8953–8966. [Google Scholar] [CrossRef] [PubMed]
- Raychoudhuri, A.; Shrivastava, S.; Steele, R.; Dash, S.; Kanda, T.; Ray, R.; Ray, R.B. Hepatitis C virus infection impairs IRF-7 translocation and A interferon synthesis in immortalized human hepatocytes. J. Virol. 2010, 84, 10991–10998. [Google Scholar] [CrossRef] [PubMed]
- Gale, M., Jr.; Korth, M.J.; Tang, N.M.; Tan, S.L.; Hopkins, D.A.; Dever, T.E.; Polyak, S.J.; Gretch, D.R.; Katze, M.G. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology 1997, 230, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Nagano-Fujii, M.; Akutsu, M.; Kadoya, H.; Ohgimoto, S.; Ishido, S.; Hotta, H. Hepatitis C virus NS5A protein interacts with 2',5'-oligoadenylate synthetase and inhibits antiviral activity of IFN in an IFN sensitivity-determining region-independent manner. J. Gen. Virol. 2004, 85, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Rolle, M.; Boer, E.F.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.J.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; et al. The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashour, J.; Morrison, J.; Laurent-Rolle, M.; Belicha-Villanueva, A.; Plumlee, C.R.; Bernal-Rubio, D.; Williams, K.L.; Harris, E.; Fernandez-Sesma, A.; Schindler, C.; et al. Mouse STAT2 restricts early dengue virus replication. Cell Host Microbe. 2010, 8, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Werme, K.; Wigerius, M.; Johansson, M. Tick-borne encephalitis virus NS5 associates with membrane protein scribble and impairs interferon-stimulated JAK-STAT signalling. Cell. Microbiol. 2008, 10, 696–712. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Chang, B.L.; Yu, H.P.; Liao, C.L.; Lin, Y.L. Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J. Virol. 2006, 80, 5908–5918. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, M.; Jones, M.; Davidson, A.; Chain, B.; Jacobs, M. Dengue virus NS5 inhibits interferon-α signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J. Infect. Dis. 2009, 200, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Szretter, K.J.; Daniels, B.P.; Cho, H.; Gainey, M.D.; Yokoyama, W.M.; Gale Jr, M.; Virgin, H.W.; Klein, R.S.; Sen, G.C.; Diamond, M.S. 2′-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and-independent mechanisms of host restriction in vivo. PLoS Pathog. 2012, 8, e1002698. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Virus | Protein | Immune Evasion Mechanisms | Reference |
|---|---|---|---|
| DENV | NS2A | Inhibit RIG-I/MAVS signaling by blocking TBK1/IRF3 phosphorylation | [51] |
| DENV | NS2B3 | Inhibit type I IFN production by cleaving human STING | [52] |
| Inhibit innate immunity by cleaving STING | [53] | ||
| HCV | NS2 | Interact with IKKε and TBK1 leading the inhibition of IRF3 phosphorylation | [54] |
| KUN | NS2A | Inhibit the induction of IFN-β promoter-driven transcription | [49] |
| Virus | Protein | Immune Evasion Mechanisms | Reference |
|---|---|---|---|
| HCV | NS3 | Interact with TBK1 leading the IRF3 inhibition | [73] |
| Interact with LUBAC leading the inhibition of NF-κB activation | [75] | ||
| HCV | NS3/4A | Inhibit TLR3 signaling pathway by cleaving TRIF | [76] |
| Block the IRF3 phosphorylation | [77] | ||
| Inhibit RLR signaling by binding and cleaving MAVS | [15,76] |
| Virus | Protein | Immune Evasion Mechanisms | Reference |
|---|---|---|---|
| DENV | NS4A | Inhibit IFNβ mediated ISRE54 promoter activation | [50] |
| HCV | NS4B | Compete with STING for binding to MAVS to influence RLR signaling | [84] |
| Compete with STING for binding to TBK1 to influence RLR signaling | [85] | ||
| Inhibit STING accumulation led the suppression of RLR signaling | [86] | ||
| WNV/DENV | NS4B | Inhibit RIG-I/MAVS signaling by blocking TBK1/IRF3 phosphorylation | [51] |
| DENV | NS4B | Inhibit IFN-mediated STAT1 phosphorylation | [50] |
| YFV/WNV/DENV | NS4B | Inhibit the activation of ISRE by IFNβ stimulation | [51,83,87] |
| Virus | Protein | Immune Evassion Mechanisms | Reference |
|---|---|---|---|
| HCV | NS5A | Impair TLR-MyD88 signaling via binding to MyD88 | [19] |
| Impair the nuclear translocation of IRF7 after MyD88 activation | [104] | ||
| Bind STAT1 to inhibit JAK/STAT signaling pathway | [87] | ||
| Bind to PKR and inhibit its activity | [110] | ||
| Inhibit 2′,5′-oligoadenylate synthetase function | [111] | ||
| WNV | NS5 | Impair IFN-mediated JAK-STAT signaling by suppressing STAT1 phosphorylation | [112] |
| DENV | NS5 | Inhibit IFN-mediated signaling by blocking STAT2 phosphorylation | [113] |
| YFV | NS5 | Inhibit type I IFN mediated signaling by binding to STAT2 | [106] |
| ZIKV | NS5 | Target STAT2 to inhibit Type I interferon signaling | [107] |
| LGTV | NS5 | Inhibit the JAK-STAT signaling by interacting with IFNAR2/IFNAR1 | [105] |
| TBEV | NS5 | Impair JAK/STAT signaling by blocking STAT1 phosphorylation | [114] |
| JEV | NS5 | Impair JAK-STAT signaling by blocking STAT1 and Tyk2 phosphorylation | [115] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Wu, Z.; Wang, M.; Cheng, A. Innate Immune Evasion Mediated by Flaviviridae Non-Structural Proteins. Viruses 2017, 9, 291. https://doi.org/10.3390/v9100291
Chen S, Wu Z, Wang M, Cheng A. Innate Immune Evasion Mediated by Flaviviridae Non-Structural Proteins. Viruses. 2017; 9(10):291. https://doi.org/10.3390/v9100291
Chicago/Turabian StyleChen, Shun, Zhen Wu, Mingshu Wang, and Anchun Cheng. 2017. "Innate Immune Evasion Mediated by Flaviviridae Non-Structural Proteins" Viruses 9, no. 10: 291. https://doi.org/10.3390/v9100291
APA StyleChen, S., Wu, Z., Wang, M., & Cheng, A. (2017). Innate Immune Evasion Mediated by Flaviviridae Non-Structural Proteins. Viruses, 9(10), 291. https://doi.org/10.3390/v9100291