
Disentangling the Frames, the State of Research on the Alphavirus 6K and TF Proteins


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Assembly-Centric Alphavirus Lifecycle
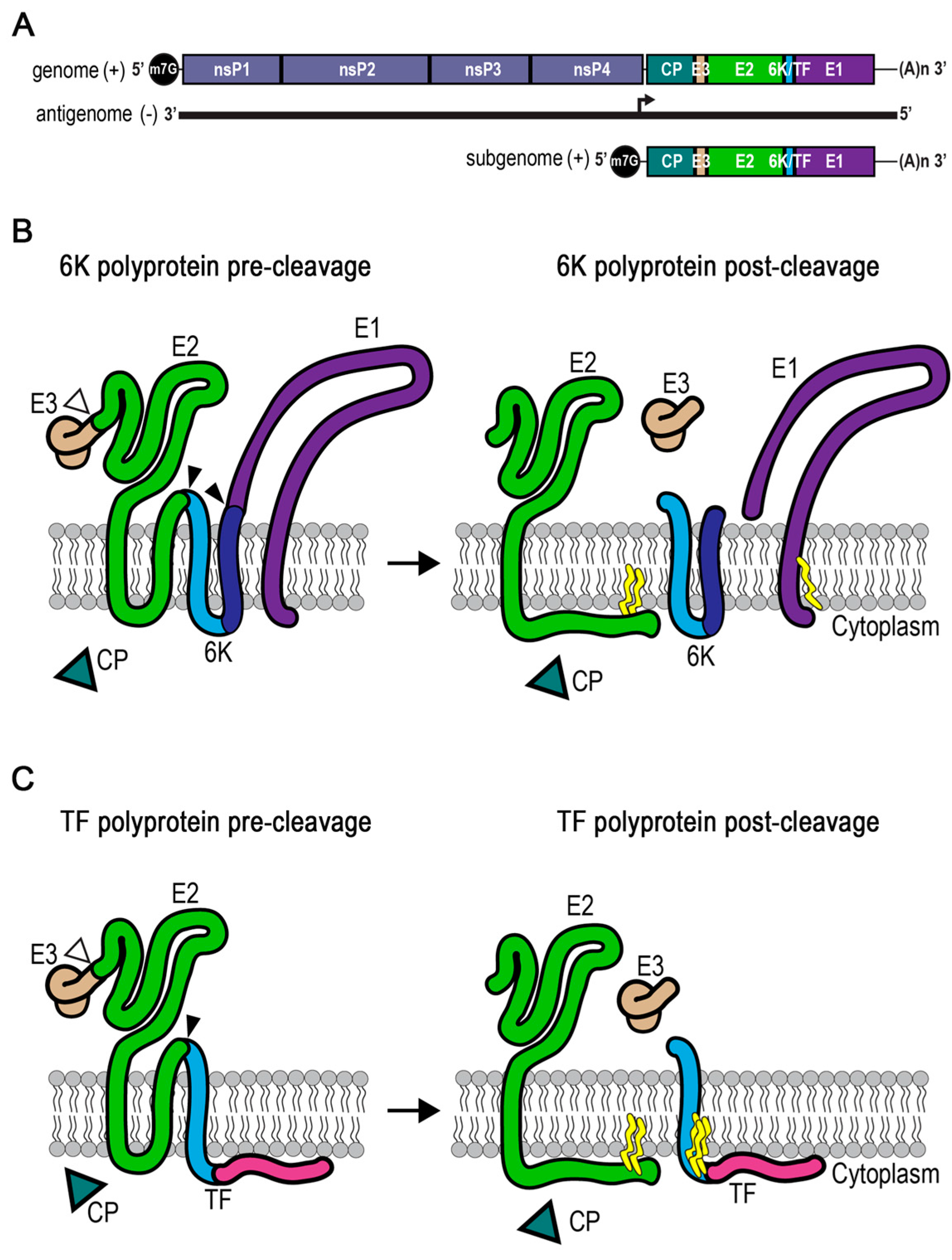
3. Enter TF, the Other 6K
4. Looking Back at the Discovery of the 6K Protein
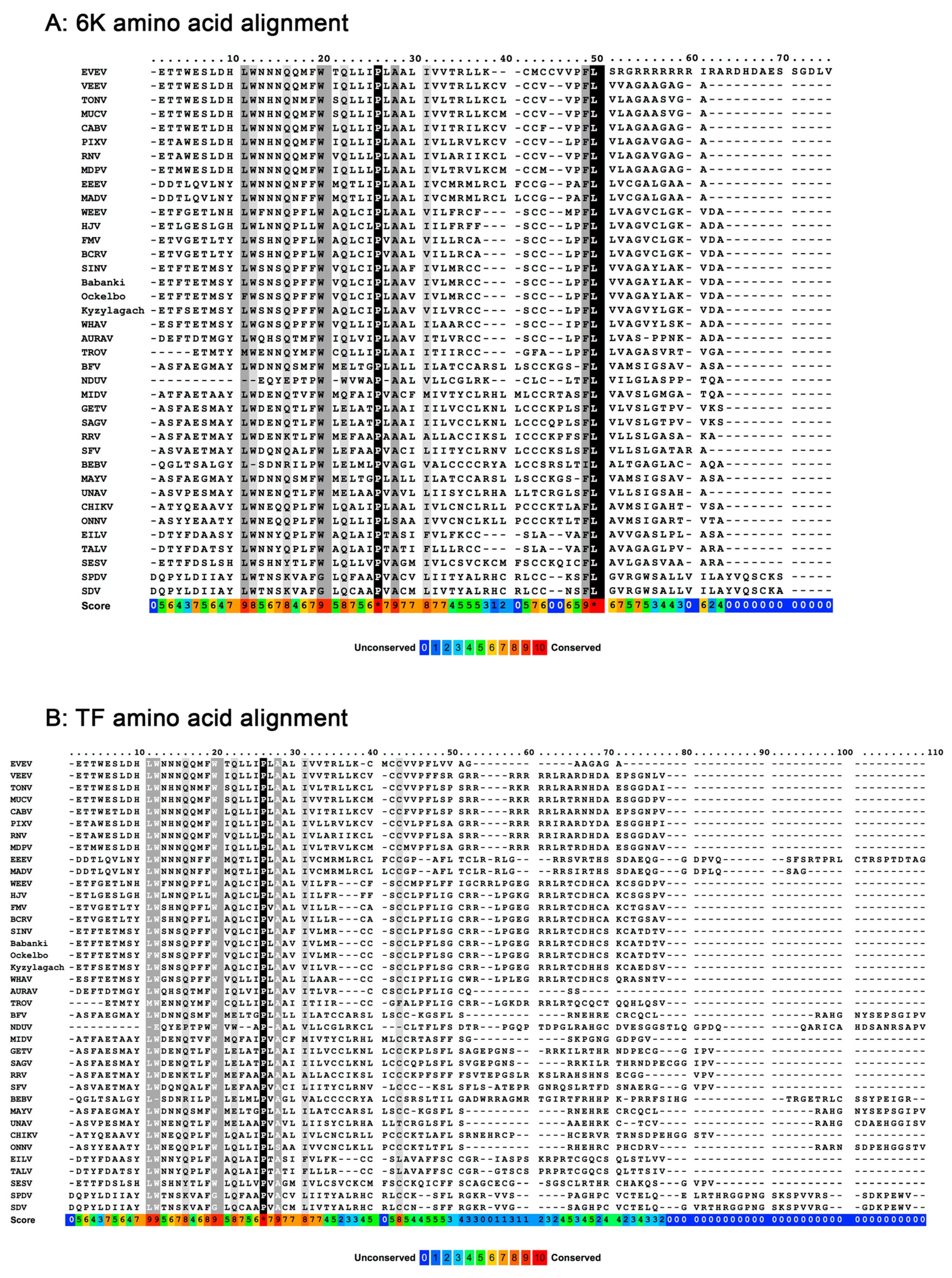
5. Posttranslational Modification in TF, but not 6K
6. Mutations that Control the Production of 6K and TF during an Infection
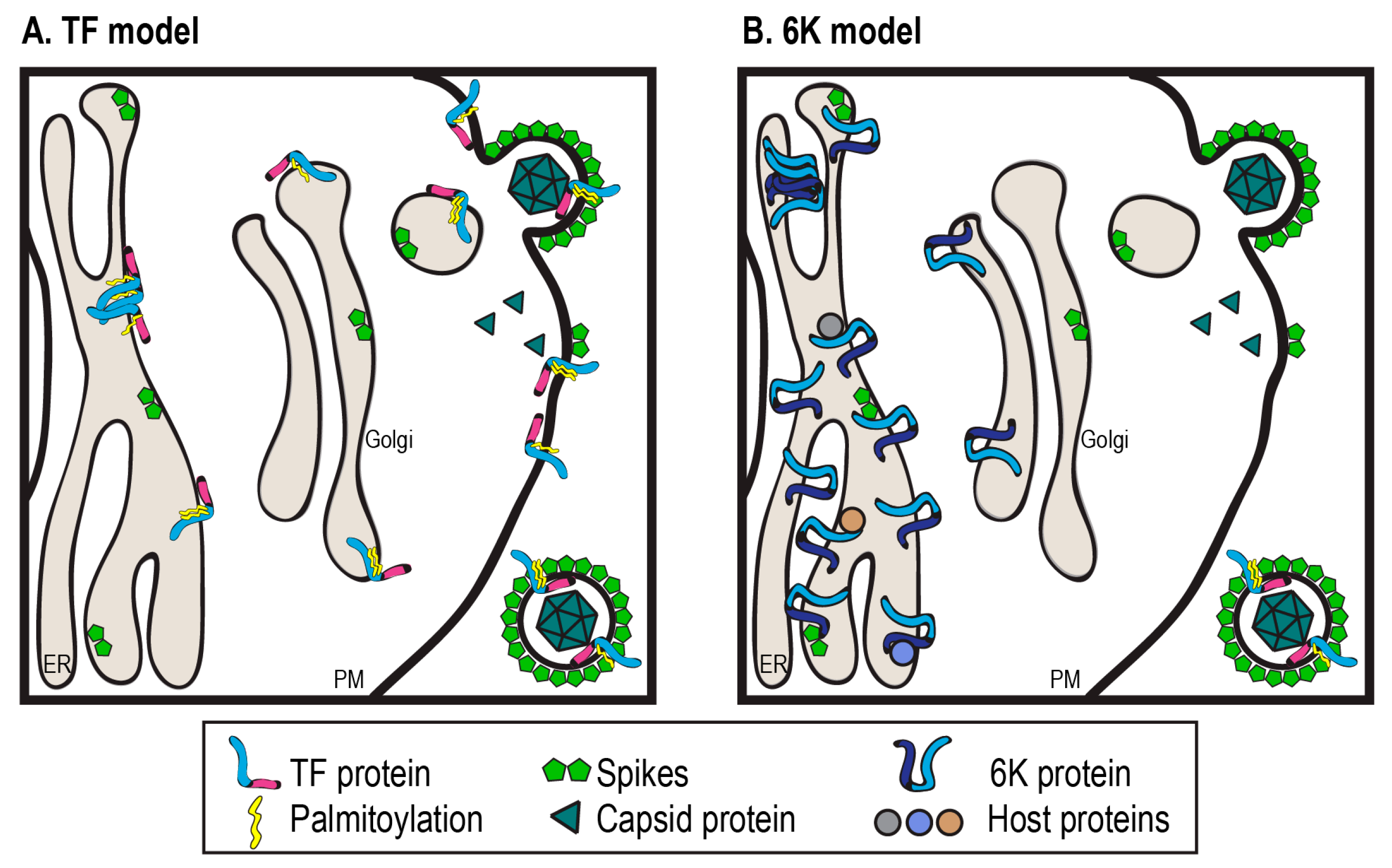
7. Evidence that 6K Functions as a Spacer during Spike Assembly
8. Direct Evidence for a 6K:E2 Interaction in Glycoprotein Maturation
9. 6K is a Viroporin
10. TF as a Virulence Determinant in Animal Infections
11. Future Directions in 6K and TF Research
11.1. How Does 6K Promote Budding from Inside the Cell?
11.2. What Regulates TF Translation and Palmitoylation?
11.3. What is the Structure and Oligomeric State of the 6K and TF Proteins?
11.4. Can We Target the Budding Proteins 6K and TF for Antivirals?
12. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kuhn, R.J. Togaviridae. In Fields Virology; Knipe, D.M., Howley, P., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Chapter 22; p. 2664. [Google Scholar]
- Sokoloski, K.J.; Snyder, A.J.; Liu, N.H.; Hayes, C.A.; Mukhopadhyay, S.; Hardy, R.W. Encapsidation of Host-Derived Factors Correlates with Enhanced Infectivity of Sindbis Virus. J. Virol. 2013, 87, 12216–12226. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.E. Alphaviruses. In Fields Virology; Howley, P., Knipe, D.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Chapter 23; Volume 1, p. 2664. [Google Scholar]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [PubMed]
- Rupp, J.C.; Sokoloski, K.J.; Gebhart, N.N.; Hardy, R.W. Alphavirus RNA synthesis and non-structural protein functions. J. Gen. Virol. 2015, 96, 2483–2500. [Google Scholar] [CrossRef] [PubMed]
- Fros, J.; Pijlman, G. Alphavirus Infection: Host Cell Shut-Off and Inhibition of Antiviral Responses. Viruses 2016, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Garmashova, N.; Gorchakov, R.; Volkova, E.; Paessler, S.; Frolova, E.; Frolov, I. The Old World and New World Alphaviruses Use Different Virus-Specific Proteins for Induction of Transcriptional Shutoff. J. Virol. 2007, 81, 2472–2484. [Google Scholar] [CrossRef] [PubMed]
- Atasheva, S.; Garmashova, N.; Frolov, I.; Frolova, E. Venezuelan Equine Encephalitis Virus Capsid Protein Inhibits Nuclear Import in Mammalian but not in Mosquito Cells. J. Virol. 2008, 82, 4028–4041. [Google Scholar] [CrossRef] [PubMed]
- Akhrymuk, I.; Kulemzin, S.V.; Frolova, E.I. Evasion of the innate immune response: The Old World alphavirus nsP2 protein induces rapid degradation of Rpb1, a catalytic subunit of RNA polymerase II. J. Virol. 2012, 86, 7180–7191. [Google Scholar] [CrossRef] [PubMed]
- Aliperti, G.; Schlesinger, M.J. Evidence for an autoprotease activity of Sindbis virus capsid protein. Virology 1978, 90, 366–369. [Google Scholar] [CrossRef]
- Melancon, P.; Garoff, H. Processing of the Semliki Forest virus structural polyprotein: Role of the capsid protease. J. Virol. 1987, 61, 1301–1309. [Google Scholar] [PubMed]
- Hahn, C.S.; Strauss, J.H. Site-directed mutagenesis of the proposed catalytic amino acids of the Sindbis virus capsid protein autoprotease. J. Virol. 1990, 64, 3069–3073. [Google Scholar] [PubMed]
- Gaedigk-Nitschko, K.; Ding, M.X.; Levy, M.A.; Schlesinger, M.J. Site-directed mutations in the Sindbis virus 6K protein reveal sites for fatty acylation and the underacylated protein affects virus release and virion structure. Virology 1990, 175, 282–291. [Google Scholar] [CrossRef]
- Firth, A.E.; Chung, B.Y.; Fleeton, M.N.; Atkins, J.F. Discovery of frameshifting in Alphavirus 6K resolves a 20-year enigma. Virol. J. 2008, 5, 108. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.E.; Kulcsar, K.A.; Schultz, K.L.W.; Riley, C.P.; Neary, J.T.; Marr, S.; Jose, J.; Griffin, D.E.; Kuhn, R.J. Functional characterization of the alphavirus TF protein. J. Virol. 2013, 87, 8511–8523. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, J.; Renzi, E.C.; Arnold, R.J.; Trinidad, J.C.; Mukhopadhyay, S. Palmitoylation of Sindbis Virus TF Protein Regulates Its Plasma Membrane Localization and Subsequent Incorporation into Virions. J. Virol. 2017, 91, e02000-16. [Google Scholar] [CrossRef] [PubMed]
- Rice, C.M.; Strauss, J.H. Nucleotide sequence of the 26S mRNA of Sindbis virus and deduced sequence of the encoded virus structural proteins. Proc. Natl. Acad. Sci. USA 1981, 78, 2062–2066. [Google Scholar] [CrossRef] [PubMed]
- Dalbey, R.E.; Lively, M.O.; Bron, S.; van Dijl, J.M. The chemistry and enzymology of the type I signal peptidases. Protein Sci. 1997, 6, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, M.; Brown, D.T. Assembly of the Sindbis virus spike protein complex. Virology 1996, 219, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Carleton, M.; Brown, D.T. Disulfide bridge-mediated folding of Sindbis virus glycoproteins. J. Virol. 1996, 70, 5541–5547. [Google Scholar] [PubMed]
- Molinari, M.; Helenius, A. Glycoproteins form mixed disulphides with oxidoreductases during folding in living cells. Nature 1999, 402, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Molinari, M. Chaperone Selection During Glycoprotein Translocation into the Endoplasmic Reticulum. Science 2000, 288, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Parrott, M.M.; Sitarski, S.A.; Arnold, R.J.; Picton, L.K.; Hill, R.B.; Mukhopadhyay, S. Role of Conserved Cysteines in the Alphavirus E3 Protein. J. Virol. 2009, 83, 2584–2591. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, M.; Brown, D.T. Involvement of the molecular chaperone BiP in maturation of Sindbis virus envelope glycoproteins. J. Virol. 1995, 69, 1621–1627. [Google Scholar] [PubMed]
- Martín, C.S.-S.; Liu, C.Y.; Kielian, M. Dealing with low pH: Entry and exit of alphaviruses and flaviviruses. Trends Microbiol. 2009, 17, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Jose, J.; Snyder, J.E.; Kuhn, R.J. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 2009, 4, 837–856. [Google Scholar] [CrossRef] [PubMed]
- Lobigs, M.; Zhao, H.X.; Garoff, H. Function of Semliki Forest virus E3 peptide in virus assembly: Replacement of E3 with an artificial signal peptide abolishes spike heterodimerization and surface expression of E1. J. Virol. 1990, 64, 4346–4355. [Google Scholar] [PubMed]
- Zhang, X.; Fugère, M.; Day, R.; Kielian, M. Furin processing and proteolytic activation of Semliki Forest virus. J. Virol. 2003, 77, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Müller, B.; Kräusslich, H.-G. More than one door—Budding of enveloped viruses through cellular membranes. FEBS Lett. 2007, 581, 2089–2097. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Sundquist, W.I. Virus Budding and the ESCRT Pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.M.; Hanson, P.I.; Kielian, M. Ubiquitin Depletion and Dominant-Negative VPS4 Inhibit Rhabdovirus Budding without Affecting Alphavirus Budding. J. Virol. 2007, 81, 13631–13639. [Google Scholar] [CrossRef] [PubMed]
- Yondola, M.; Carter, C. Un-“ESCRT-”ed Budding. Viruses 2011, 3, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, T.A.; Tellinghuisen, T.L.; Kuhn, R.J.; Post, C.B. Association of Sindbis Virus Capsid Protein with Phospholipid Membranes and the E2 Glycoprotein: Implications for Alphavirus Assembly. Biochemistry 2005, 44, 2800–2810. [Google Scholar] [CrossRef] [PubMed]
- Skoging, U.; Vihinen, M.; Nilsson, L.; Liljeström, P. Aromatic interactions define the binding of the alphavirus spike to its nucleocapsid. Structure 1996, 4, 519–529. [Google Scholar] [CrossRef]
- Lee, S.; Owen, K.E.; Choi, H.K.; Lee, H.; Lu, G.; Wengler, G.; Brown, D.T.; Rossmann, M.G.; Kuhn, R.J. Identification of a protein binding site on the surface of the alphavirus nucleocapsid and its implication in virus assembly. Structure 1996, 4, 531–541. [Google Scholar] [CrossRef]
- Soonsawad, P.; Xing, L.; Milla, E.; Espinoza, J.M.; Kawano, M.; Marko, M.; Hsieh, C.; Furukawa, H.; Kawasaki, M.; Weerachatyanukul, W.; et al. Structural Evidence of Glycoprotein Assembly in Cellular Membrane Compartments prior to Alphavirus Budding. J. Virol. 2010, 84, 11145–11151. [Google Scholar] [CrossRef] [PubMed]
- Jose, J.; Taylor, A.B.; Kuhn, R.J. Spatial and Temporal Analysis of Alphavirus Replication and Assembly in Mammalian and Mosquito Cells. mBio 2017, 8, e02294-16. [Google Scholar] [CrossRef] [PubMed]
- Acheson, N.H.; Tamm, I. Replication of Semliki Forest virus: An electron microscopic study. Virology 1967, 32, 128–143. [Google Scholar] [CrossRef]
- Miller, M.L.; Brown, D.T. Morphogenesis of Sindbis virus in three subclones of Aedes albopictus (mosquito) cells. J. Virol. 1992, 66, 4180–4190. [Google Scholar] [PubMed]
- Forrester, N.L.; Palacios, G.; Tesh, R.B.; Savji, N.; Guzman, H.; Sherman, M.; Weaver, S.C.; Lipkin, W.I. Genome-Scale Phylogeny of the Alphavirus Genus Suggests a Marine Origin. J. Virol. 2012, 86, 2729–2738. [Google Scholar] [CrossRef] [PubMed]
- Nasar, F.; Palacios, G.; Gorchakov, R.V.; Guzman, H.; Da Rosa, A.P.T.; Savji, N.; Popov, V.L.; Sherman, M.B.; Lipkin, W.I.; Tesh, R.B. Eilat virus, a unique alphavirus with host range restricted to insects by RNA replication. Proc. Natl. Acad. Sci. USA 2012, 109, 14622–14627. [Google Scholar] [CrossRef] [PubMed]
- Hermanns, K.; Zirkel, F.; Kopp, A.; Marklewitz, M.; Rwego, I.B.; Estrada, A.; Gillespie, T.R.; Drosten, C.; Junglen, S. Discovery of a novel alphavirus related to Eilat virus. J. Gen. Virol. 2017, 98, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.-C.; Johansson, D.X.; Haugland, Ø.; Liljeström, P.; Evensen, Ø. A 6K-Deletion Variant of Salmonid Alphavirus Is Non-Viable but Can Be Rescued through RNA Recombination. PLoS ONE 2014, 9, e100184. [Google Scholar] [CrossRef] [PubMed]
- Forrester, N.L.; Guerbois, M.; Adams, A.P.; Liang, X.; Weaver, S.C. Analysis of Intrahost Variation in Venezuelan Equine Encephalitis Virus Reveals Repeated Deletions in the 6-Kilodalton Protein Gene. J. Virol. 2011, 85, 8709–8717. [Google Scholar] [CrossRef] [PubMed]
- Petterson, E.; Stormoen, M.; Evensen, O.; Mikalsen, A.B.; Haugland, O. Natural infection of Atlantic salmon (Salmo salar L.) with salmonid alphavirus 3 (SAV3) generates numerous viral deletion mutants. J. Gen. Virol. 2013, 94, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Petterson, E.; Guo, T.-C.; Evensen, Ø.; Mikalsen, A.B. Experimental piscine alphavirus RNA recombination in vivo yields both viable virus and defective viral RNA. Sci. Rep. 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Power, M.D.; Masiarz, F.R.; Luciw, P.A.; Barr, P.J.; Varmus, H.E. Characterization of ribosomal frameshifting in HIV-1 Gag-Pol expression. Nature 1988, 331, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Madhani, H.D.; Masiarz, F.R.; Varmus, H.E. Signals for ribosomal frameshifting in the Rous sarcoma virus Gag-Pol region. Cell 1988, 55, 447–458. [Google Scholar] [CrossRef]
- Jacks, T.; Varmus, H.E. Expression of the Rous sarcoma virus Pol gene by ribosomal frameshifting. Science 1985, 230, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Dinman, J.D. Mechanisms and implications of programmed translational frameshifting. WIREs RNA 2012, 3, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Atkins, J.F.; Loughran, G.; Bhatt, P.R.; Firth, A.E.; Baranov, P.V. Ribosomal frameshifting and transcriptional slippage: From genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 2016, 44, 7007–7078. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.Y.W.; Firth, A.E.; Atkins, J.F. Frameshifting in Alphaviruses: A Diversity of 3’ Stimulatory Structures. J. Mol. Biol. 2010, 397, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Kendra, J.A.; de la Fuente, C.; Brahms, A.; Woodson, C.; Bell, T.M.; Chen, B.; Khan, Y.A.; Jacobs, J.L.; Kehn-Hall, K.; Dinman, J.D. Ablation of Programmed -1 Ribosomal Frameshifting in Venezuelan Equine Encephalitis Virus Results in Attenuated Neuropathogenicity. J. Virol. 2017, 91, e01766-16. [Google Scholar] [CrossRef] [PubMed]
- Welch, W.J.; Sefton, B.M. Two small virus-specific polypeptides are produced during infection with Sindbis virus. J. Virol. 1979, 29, 1186–1195. [Google Scholar] [PubMed]
- Gaedigk-Nitschko, K.; Schlesinger, M.J. Site-directed mutations in Sindbis virus E2 glycoprotein’s cytoplasmic domain and the 6K protein lead to similar defects in virus assembly and budding. Virology 1991, 183, 206–214. [Google Scholar] [CrossRef]
- Melton, J.V.; Ewart, G.D.; Weir, R.C.; Board, P.G.; Lee, E.; Gage, P.W. Alphavirus 6K Proteins Form Ion Channels. J. Biol. Chem. 2002, 277, 46923–46931. [Google Scholar] [CrossRef] [PubMed]
- International Committee on the Taxonomy of Viruses. Available online: https://talk.ictvonline.org/ (accessed on 26 May 2017).
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [PubMed]
- Heringa, J. Two strategies for sequence comparison: Profile-preprocessed and secondary structure-induced multiple alignment. Comput. Chem. 1999, 23, 341–364. [Google Scholar] [CrossRef]
- Simossis, V.A.; Heringa, J. The PRALINE online server: Optimising progressive multiple alignment on the web. Comput. Biol. Chem. 2003, 27, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Simossis, V.A.; Kleinjung, J.; Heringa, J. Homology-extended sequence alignment. Nucleic Acids Res. 2005, 33, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Simossis, V.A.; Heringa, J. PRALINE: A multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res. 2005, 33, W289–W294. [Google Scholar] [CrossRef] [PubMed]
- Liljeström, P.; Lusa, S.; Huylebroeck, D.; Garoff, H. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: The small 6000-molecular-weight membrane protein modulates virus release. J. Virol. 1991, 65, 4107–4113. [Google Scholar] [PubMed]
- Loewy, A.; Smyth, J.; von Bonsdorff, C.H.; Liljeström, P.; Schlesinger, M.J. The 6-kilodalton membrane protein of Semliki Forest virus is involved in the budding process. J. Virol. 1995, 69, 469–475. [Google Scholar] [PubMed]
- Ivanova, L.; Lustig, S.; Schlesinger, M.J. A pseudo-revertant of a Sindbis virus 6K protein mutant, which corrects for aberrant particle formation, contains two new mutations that map to the ectodomain of the E2 glycoprotein. Virology 1995, 206, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Melton, J.V.; Herrero, L.J.; Thaa, B.; Karo-Astover, L.; Gage, P.W.; Nelson, M.A.; Sheng, K.-C.; Lidbury, B.A.; Ewart, G.D.; et al. Effects of an in-frame deletion of the 6K gene locus from the genome of Ross River virus. J. Virol. 2016, 90, 4150–4159. [Google Scholar] [CrossRef] [PubMed]
- Bonatti, S.; Blobel, G. Absence of a cleavable signal sequence in Sindbis virus glycoprotein PE2. J. Biol. Chem. 1979, 254, 12261–12264. [Google Scholar] [PubMed]
- Bonatti, S.; Migliaccio, G.; Blobel, G.; Walter, P. Role of signal recognition particle in the membrane assembly of Sindbis viral glycoproteins. Eur. J. Biochem. 1984, 140, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Garoff, H.; Huylebroeck, D.; Robinson, A.; Tillman, U.; Liljeström, P. The signal sequence of the p62 protein of Semliki Forest virus is involved in initiation but not in completing chain translocation. J. Cell Biol. 1990, 111, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Burge, B.W.; Strauss, J.H. Glycopeptides of the membrane glycoprotein of Sindbis virus. J. Mol. Biol. 1970, 47, 449–466. [Google Scholar] [CrossRef]
- Rice, C.M.; Bell, J.R.; Hunkapiller, M.W.; Strauss, E.G.; Strauss, J.H. Isolation and characterization of the hydrophobic COOH-terminal domains of the sindbis virion glycoproteins. J. Mol. Biol. 1982, 154, 355–378. [Google Scholar] [CrossRef]
- Welch, W.J.; Sefton, B.M. Characterization of a small, nonstructural viral polypeptide present late during infection of BHK cells by Semliki Forest virus. J. Virol. 1980, 33, 230–237. [Google Scholar] [PubMed]
- Welch, W.J.; Sefton, B.M.; Esch, F.S. Amino-terminal sequence analysis of alphavirus polypeptides. J. Virol. 1981, 38, 968–972. [Google Scholar] [PubMed]
- Rice, C.M.; Strauss, J.H. Synthesis, cleavage and sequence analysis of DNA complementary to the 26 S messenger RNA of Sindbis virus. J. Mol. Biol. 1981, 150, 315–340. [Google Scholar] [CrossRef]
- Hashimoto, K.; Erdei, S.; Keränen, S.; Saraste, J.; Kaariainen, L. Evidence for a separate signal sequence for the carboxy-terminal envelope glycoprotein E1 of Semliki forest virus. J. Virol. 1981, 38, 34–40. [Google Scholar] [PubMed]
- Melancon, P.; Garoff, H. Reinitiation of translocation in the Semliki Forest virus structural polyprotein: Identification of the signal for the E1 glycoprotein. EMBO J. 1986, 5, 1551–1560. [Google Scholar] [PubMed]
- Liljeström, P.; Garoff, H. Internally located cleavable signal sequences direct the formation of Semliki Forest virus membrane proteins from a polyprotein precursor. J. Virol. 1991, 65, 147–154. [Google Scholar] [PubMed]
- Migliaccio, G.; Pascale, M.C.; Leone, A.; Bonatti, S. Biosynthesis, membrane translocation, and surface expression of Sindbis virus E1 glycoprotein. Exp. Cell Res. 1989, 185, 203–216. [Google Scholar] [CrossRef]
- Metz, S.W.; Geertsema, C.; Martina, B.E.; Andrade, P.; Heldens, J.G.; Van Oers, M.M.; Goldbach, R.W.; Vlak, J.M.; Pijlman, G.P. Functional processing and secretion of Chikungunya virus E1 and E2 glycoproteins in insect cells. Virol. J. 2011, 8, 353. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.F.; Bracha, M.; Schlesinger, M.J. Evidence for covalent attachment of fatty acids to Sindbis virus glycoproteins. Proc. Natl. Acad. Sci. USA 1979, 76, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.F.; Schlesinger, M.J. Fatty acid binding to vesicular stomatitis virus glycoprotein: A new type of post-translational modification of the viral glycoprotein. Cell 1979, 17, 813–819. [Google Scholar] [CrossRef]
- Schmidt, M.F. Acylation of viral spike glycoproteins: A feature of enveloped RNA viruses. Virology 1982, 116, 327–338. [Google Scholar] [CrossRef]
- Schmidt, M.; Schmidt, M.F.; Rott, R. Chemical identification of cysteine as palmitoylation site in a transmembrane protein (Semliki Forest virus E1). J. Biol. Chem. 1988, 263, 18635–18639. [Google Scholar] [PubMed]
- Ivanova, L.; Schlesinger, M.J. Site-directed mutations in the Sindbis virus E2 glycoprotein identify palmitoylation sites and affect virus budding. J. Virol. 1993, 67, 2546–2551. [Google Scholar] [PubMed]
- Ryan, C.; Ivanova, L.; Schlesinger, M.J. Effects of Site-Directed Mutations of Transmembrane Cysteines in Sindbis Virus E1 and E2 Glycoproteins on Palmitylation and Virus Replication. Virology 1998, 249, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Gaedigk-Nitschko, K.; Schlesinger, M.J. The Sindbis virus 6K protein can be detected in virions and is acylated with fatty acids. Virology 1990, 175, 274–281. [Google Scholar] [CrossRef]
- McInerney, G.M.; Smit, J.M.; Liljeström, P.; Wilschut, J. Semliki Forest virus produced in the absence of the 6K protein has an altered spike structure as revealed by decreased membrane fusion capacity. Virology 2004, 325, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Carrasco, L. Sindbis virus variant with a deletion in the 6K gene shows defects in glycoprotein processing and trafficking: Lack of complementation by a wild-type 6K gene in trans. J. Virol. 2001, 75, 7778–7784. [Google Scholar] [CrossRef] [PubMed]
- Metz, S.W.; Feenstra, F.; Villoing, S.; van Hulten, M.C.; van Lent, J.W.; Koumans, J.; Vlak, J.M.; Pijlman, G.P. Low Temperature-Dependent Salmonid Alphavirus Glycoprotein Processing and Recombinant Virus-Like Particle Formation. PLoS ONE 2011, 6, e25816. [Google Scholar] [CrossRef] [PubMed]
- Hikke, M.C.; Braaen, S.; Villoing, S.; Hodneland, K.; Geertsema, C.; Verhagen, L.; Frost, P.; Vlak, J.M.; Rimstad, E.; Pijlman, G.P. Salmonid alphavirus glycoprotein E2 requires low temperature and E1 for virion formation and induction of protective immunity. Vaccine 2014, 32, 6206–6212. [Google Scholar] [CrossRef] [PubMed]
- Rice, C.M.; Levis, R.; Strauss, J.H.; Huang, H.V. Production of infectious RNA transcripts from Sindbis virus cDNA clones: Mapping of lethal mutations, rescue of a temperature-sensitive marker, and in vitro mutagenesis to generate defined mutants. J. Virol. 1987, 61, 3809–3819. [Google Scholar] [PubMed]
- Ivanova, L.; Le, L.; Schlesinger, M.J. Characterization of revertants of a Sindbis virus 6K gene mutant that affects proteolytic processing and virus assembly. Virus Res. 1995, 39, 165–179. [Google Scholar] [CrossRef]
- Strauss, E.G.; Birdwell, C.R.; Lenches, E.M.; Staples, S.E.; Strauss, J.H. Mutants of Sindbis virus II. Characterization of a maturation-defective mutant, ts103. Virology 1977, 82, 122–149. [Google Scholar] [CrossRef]
- Lusa, S.; Garoff, H.; Liljeström, P. Fate of the 6K membrane protein of Semliki Forest virus during virus assembly. Virology 1991, 185, 843–846. [Google Scholar] [CrossRef]
- Wu, D.; Xu, W.; Jiao, R.; Ding, M.; Zhai, Z. The relationship of Sindbis virus assembly and the viral protein 6K with intermediate filaments. Acta Microbiol. Sin. 1990, 3, 417–421. [Google Scholar]
- Yao, J.S.; Strauss, E.G.; Strauss, J.H. Interactions between PE2, E1, and 6K required for assembly of alphaviruses studied with chimeric viruses. J. Virol. 1996, 70, 7910–7920. [Google Scholar] [PubMed]
- Snyder, A.J.; Sokoloski, K.J.; Mukhopadhyay, S. Mutating conserved cysteines in the alphavirus E2 glycoprotein causes virus-specific assembly defects. J. Virol. 2012, 86, 3100–3111. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.J.; Mukhopadhyay, S. The Alphavirus E3 Glycoprotein Functions in a Clade-Specific Manner. J. Virol. 2012, 86, 13609–13620. [Google Scholar] [CrossRef] [PubMed]
- Strauss, E.G.; Lenches, E.M.; Strauss, J.H. Molecular Genetic Evidence that the Hydrophobic Anchors of Glycoproteins E2 and E1 Interact during Assembly of Alphaviruses. J. Virol. 2002, 76, 10188–10194. [Google Scholar] [CrossRef]
- London, S.D.; Schmaljohn, A.L.; Dalrymple, J.M.; Rice, C.M. Infectious enveloped RNA virus antigenic chimeras. Proc. Natl. Acad. Sci. USA 1992, 89, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, M.J.; London, S.D.; Ryan, C. An in-frame insertion into the Sindbis virus 6K gene leads to defective proteolytic processing of the virus glycoproteins, a trans-dominant negative inhibition of normal virus formation, and interference in virus shut off of host-cell protein synthesis. Virology 1993, 193, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.B.; Krüger, J. Viral Channel-Forming Proteins. Int. Rev. Cell Mol. Biol. 2009, 275, 35–63. [Google Scholar] [PubMed]
- Martinez-Gil, L.; Mingarro, I. Viroporins, Examples of the Two-Stage Membrane Protein Folding Model. Viruses 2015, 7, 3462–3482. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, L. Modification of membrane permeability by animal viruses. Adv. Virus Res. 1995. [Google Scholar] [CrossRef]
- Sanz, M.A.; Pérez, L.; Carrasco, L. Semliki Forest virus 6K protein modifies membrane permeability after inducible expression in Escherichia coli cells. J. Biol. Chem. 1994, 269, 12106–12110. [Google Scholar] [PubMed]
- Sanz, M.A.; Madan, V.; Carrasco, L.; Nieva, J.L. Interfacial domains in Sindbis virus 6K protein. Detection and functional characterization. J. Biol. Chem. 2003, 278, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Sanz, M.A.; Carrasco, L. Requirement of the vesicular system for membrane permeabilization by Sindbis virus. Virology 2005, 332, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; de García, M.J.; Sanz, M.A.; Carrasco, L. Viroporin activity of murine hepatitis virus E protein. FEBS Lett. 2005, 579, 3607–3612. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Redondo, N.; Carrasco, L. Cell permeabilization by poliovirus 2B viroporin triggers bystander permeabilization in neighbouring cells through a mechanism involving gap junctions. Cell. Microbiol. 2010, 12, 1144–1157. [Google Scholar] [CrossRef] [PubMed]
- Antoine, A.-F.; Montpellier, C.; Cailliau, K.; Browaeys-Poly, E.; Vilain, J.-P.; Dubuisson, J. The Alphavirus 6K Protein Activates Endogenous Ionic Conductances when Expressed in Xenopus Oocytes. J. Membr. Biol. 2007, 215, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.L.; Cook, J.A.; Brown-Augsburger, P.; Heinz, B.A.; Smith, M.C.; Pinto, L.H. Demonstrating the intrinsic ion channel activity of virally encoded proteins. FEBS Lett. 2003, 552, 61–67. [Google Scholar] [CrossRef]
- Jose, J.; Kuhn, R.J.; Purdue University, West Lafayette, IN, USA. Personal communication, 2017.
- Nieva, J.L.; Sanz, M.A.; Carrasco, L. Membrane-permeabilizing motif in Semliki forest virus E1 glycoprotein. FEBS Lett. 2004, 576, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.-C.; Jin, Y.; Zhi, X.-Y.; Yan, D.; Sun, S.-Q. NLRP3 Inflammasome Activation by Viroporins of Animal Viruses. Viruses 2015, 7, 3380–3391. [Google Scholar] [CrossRef] [PubMed]
- Hyser, J.M.; Estes, M.K. Pathophysiological Consequences of Calcium-Conducting Viroporins. Annu. Rev. Virol. 2015, 2, 473–496. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.; Torres, J.; Liu, D. The Emerging Roles of Viroporins in ER Stress Response and Autophagy Induction during Virus Infection. Viruses 2015, 7, 2834–2857. [Google Scholar] [CrossRef] [PubMed]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Balannik, V.; Pinto, L.H.; Lamb, R.A. Influenza Virus M2 Ion Channel Protein Is Necessary for Filamentous Virion Formation. J. Virol. 2010, 84, 5078–5088. [Google Scholar] [CrossRef] [PubMed]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Lamb, R.A. Influenza Virus M2 Protein Mediates ESCRT-Independent Membrane Scission. Cell 2010, 142, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Rossman, J.S.; Lamb, R.A. Influenza virus assembly and budding. Virology 2011, 411, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Stouffer, A.L.; Acharya, R.; Salom, D.; Levine, A.S.; di Costanzo, L.; Soto, C.S.; Tereshko, V.; Nanda, V.; Stayrook, S.; DeGrado, W.F. Structural basis for the function and inhibition of an influenza virus proton channel. Nature 2008, 451, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Schnell, J.R.; Chou, J.J. Structure and mechanism of the M2 proton channel of influenza a virus. Nature 2008, 451, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Cady, S.D.; Mishanina, T.V.; Hong, M. Structure of amantadine-bound M2 transmembrane peptide of influenza A in lipid bilayers from magic-angle-spinning solid-state NMR: The role of Ser31 in amantadine binding. J. Mol. Biol. 2009, 385, 1127–1141. [Google Scholar] [CrossRef] [PubMed]
- Luik, P.; Chew, C.; Aittoniemi, J.; Chang, J.; Wentworth, P.; Dwek, R.A.; Biggin, P.C.; Vénien-Bryan, C.; Zitzmann, N. The 3-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 12712–12716. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Bartenschlager, R. Structural and Functional Properties of the Hepatitis C Virus p7 Viroporin. Viruses 2015, 7, 4461–4481. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, A.L.; Griffin, S.; Rowlands, D.; Harris, M.; Yi, M.; Lemon, S.M.; Weinman, S.A. Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog. 2010, 6, e1001087. [Google Scholar] [CrossRef] [PubMed]
- Atoom, A.M.; Jones, D.M.; Russell, R.S. Evidence suggesting that HCV p7 protects E2 glycoprotein from premature degradation during virus production. Virus Res. 2013, 176, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Scull, M.A.; Schneider, W.M.; Flatley, B.R.; Hayden, R.; Fung, C.; Jones, C.T.; van de Belt, M.; Penin, F.; Rice, C.M. The N-terminal Helical Region of the Hepatitis C Virus p7 Ion Channel Protein Is Critical for Infectious Virus Production. PLoS Pathog. 2015, 11, e1005297. [Google Scholar] [CrossRef] [PubMed]
- Pielak, R.M.; Chou, J.J. Flu channel drug resistance: A tale of two sites. Protein Cell 2010, 1, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Aramini, J.M.; Ma, L.-C.; Krug, R.M.; Arnold, E. Structures of influenza a proteins and insights into antiviral drug targets. Nat. Struct. Mol. Biol. 2010, 17, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Biotron Limited. Available online: www.biotron.com.au (accessed 26 May 2017). (Universal Trial Number U1111-1150-4404).
- Sugrue, R.J.; Belshe, R.B.; Hay, A.J. Palmitoylation of the influenza a virus M2 protein. Virology 1990, 179, 51–56. [Google Scholar] [CrossRef]
- Veit, M.; Klenk, H.D.; Kendal, A.; Rott, R. The M2 protein of influenza a virus is acylated. J. Gen. Virol. 1991, 72(Pt. 6), 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Corse, E.; Machamer, C.E. The Cytoplasmic Tail of Infectious Bronchitis Virus E Protein Directs Golgi Targeting. J. Virol. 2002, 76, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Shipston, M.J. Ion channel regulation by protein S-acylation. J. Gen. Physiol. 2014, 143, 659–678. [Google Scholar] [CrossRef] [PubMed]
- González, M.E.; Carrasco, L. Human immunodeficiency virus type 1 VPU protein affects Sindbis virus glycoprotein processing and enhances membrane permeabilization. Virology 2001, 279, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Hallengärd, D.; Kakoulidou, M.; Lulla, A.; Kümmerer, B.M.; Johansson, D.X.; Mutso, M.; Lulla, V.; Fazakerley, J.K.; Roques, P.; Le Grand, R.; et al. Novel attenuated Chikungunya vaccine candidates elicit protective immunity in C57BL/6 mice. J. Virol. 2014, 88, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.D.; Bang Jensen, B.; McLoughlin, M.F.; Rodger, H.D.; Taksdal, T.; Sindre, H.; Graham, D.A.; Lillehaug, A. The epidemiology of pancreas disease in salmonid aquaculture: A summary of the current state of knowledge. J. Fish Dis. 2016, 40, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Weston, J.H.; Welsh, M.D.; McLoughlin, M.F.; Todd, D. Salmon pancreas disease virus, an alphavirus infecting farmed Atlantic salmon, Salmo salar L. Virology 1999, 256, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Weston, J.; Villoing, S.; Bremont, M.; Castric, J.; Pfeffer, M.; Jewhurst, V.; McLoughlin, M.; Rodseth, O.; Christie, K.E.; Koumans, J.; et al. Comparison of Two Aquatic Alphaviruses, Salmon Pancreas Disease Virus and Sleeping Disease Virus, by Using Genome Sequence Analysis, Monoclonal Reactivity, and Cross-Infection. J. Virol. 2002, 76, 6155–6163. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.D.; Gjerset, B.; Modahl, I.; Bohlin, J. Molecular epidemiology of salmonid alphavirus (SAV) subtype 3 in Norway. Virol. J. 2010, 7, 188. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Quiñonez, J.A.; Escobar-Escamilla, N.; Ortíz-Alcántara, J.; Vázquez-Pichardo, M.; Torres-Rodríguez, M.L.; Nuñez-León, A.; Torres-Longoria, B.; López-Martínez, I.; Ruiz-Matus, C.; Kuri-Morales, P.; et al. Identification of Asian genotype of chikungunya virus isolated in Mexico. Virus Genes 2016, 52, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Rodas, J.D.; Kautz, T.; Camacho, E.; Paternina, L.; Guzman, H.; Díaz, F.J.; Blanco, P.; Tesh, R.; Weaver, S.C. Genetic Characterization of Northwestern Colombian Chikungunya Virus Strains from the 2014–2015 Epidemic. Am. J. Trop. Med. Hyg. 2016, 95, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Erasmus, J.H.; Rossi, S.L.; Weaver, S.C. Development of Vaccines for Chikungunya Fever. J. Infect. Dis. 2016, 214, S488–S496. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Osorio, J.E.; Livengood, J.A.; Chen, R.; Stinchcomb, D.T. Chikungunya virus and prospects for a vaccine. Expert Rev. Vaccines 2014, 11, 1087–1101. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.H.; Kuhn, R.J.; Olson, N.H.; Rossmann, M.G.; Choi, H.K.; Smith, T.J.; Baker, T.S. Nucleocapsid and glycoprotein organization in an enveloped virus. Cell 1995, 80, 621–630. [Google Scholar] [CrossRef]
- Akahata, W.; Yang, Z.-Y.; Andersen, H.; Sun, S.; Holdaway, H.A.; Kong, W.-P.; Lewis, M.G.; Higgs, S.; Rossmann, M.G.; Rao, S.; et al. A virus-like particle vaccine for epidemic Chikungunya virus protects nonhuman primates against infection. Nat. Med. 2010, 16, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Kostyuchenko, V.A.; Jakana, J.; Liu, X.; Haddow, A.D.; Aung, M.; Weaver, S.C.; Chiu, W.; Lok, S.M. The Structure of Barmah Forest Virus as Revealed by Cryo-Electron Microscopy at a 6-Angstrom Resolution Has Detailed Transmembrane Protein Architecture and Interactions. J. Virol. 2011, 85, 9327–9333. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Zhang, W.; Gabler, S.; Chipman, P.R.; Strauss, E.G.; Strauss, J.H.; Baker, T.S.; Kuhn, R.J.; Rossmann, M.G. Mapping the Structure and Function of the E1 and E2 Glycoproteins in Alphaviruses. Structure 2006, 14, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Jose, J.; Chipman, P.; Zhang, W.; Kuhn, R.J.; Baker, T.S. Molecular Links between the E2 Envelope Glycoprotein and Nucleocapsid Core in Sindbis Virus. J. Mol. Biol. 2011, 414, 442–459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Hryc, C.F.; Cong, Y.; Liu, X.; Jakana, J.; Gorchakov, R.; Baker, M.L.; Weaver, S.C.; Chiu, W. 4.4 Å cryo-EM structure of an enveloped alphavirus Venezuelan equine encephalitis virus. EMBO J. 2011, 30, 3854–3863. [Google Scholar] [CrossRef] [PubMed]
- Ruch, T.R.; Machamer, C.E. A Single Polar Residue and Distinct Membrane Topologies Impact the Function of the Infectious Bronchitis Coronavirus E Protein. PLoS Pathog. 2012, 8, e1002674. [Google Scholar] [CrossRef] [PubMed]
- Ruch, T.R.; Machamer, C.E. The Coronavirus E Protein: Assembly and Beyond. Viruses 2012, 4, 363–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerbeck, J.W.; Machamer, C.E. A Coronavirus E Protein Is Present in Two Distinct Pools with Different Effects on Assembly and the Secretory Pathway. J. Virol. 2015, 89, 9313–9323. [Google Scholar] [CrossRef] [PubMed]
- Holodniy, M.; Kamali, A. Influenza treatment and prophylaxis with neuraminidase inhibitors: A review. Infect. Drug Resistance 2013, 6, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Von Itzstein, M.; Wu, W.Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; Van Phan, T.; Smythe, M.L.; White, H.F.; Oliver, S.W. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramsey, J.; Mukhopadhyay, S. Disentangling the Frames, the State of Research on the Alphavirus 6K and TF Proteins. Viruses 2017, 9, 228. https://doi.org/10.3390/v9080228
Ramsey J, Mukhopadhyay S. Disentangling the Frames, the State of Research on the Alphavirus 6K and TF Proteins. Viruses. 2017; 9(8):228. https://doi.org/10.3390/v9080228
Chicago/Turabian StyleRamsey, Jolene, and Suchetana Mukhopadhyay. 2017. "Disentangling the Frames, the State of Research on the Alphavirus 6K and TF Proteins" Viruses 9, no. 8: 228. https://doi.org/10.3390/v9080228
APA StyleRamsey, J., & Mukhopadhyay, S. (2017). Disentangling the Frames, the State of Research on the Alphavirus 6K and TF Proteins. Viruses, 9(8), 228. https://doi.org/10.3390/v9080228