A New Approach to Assessing HSV-1 Recombination during Intercellular Spread


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Construction of FP-Expressing Viruses
2.3. Microscopy
2.4. Quantification of Recombination
2.5. Compartmentalized Neuronal Cultures
2.6. Intravitreal Inoculation and Tissue Harvesting
3. Results
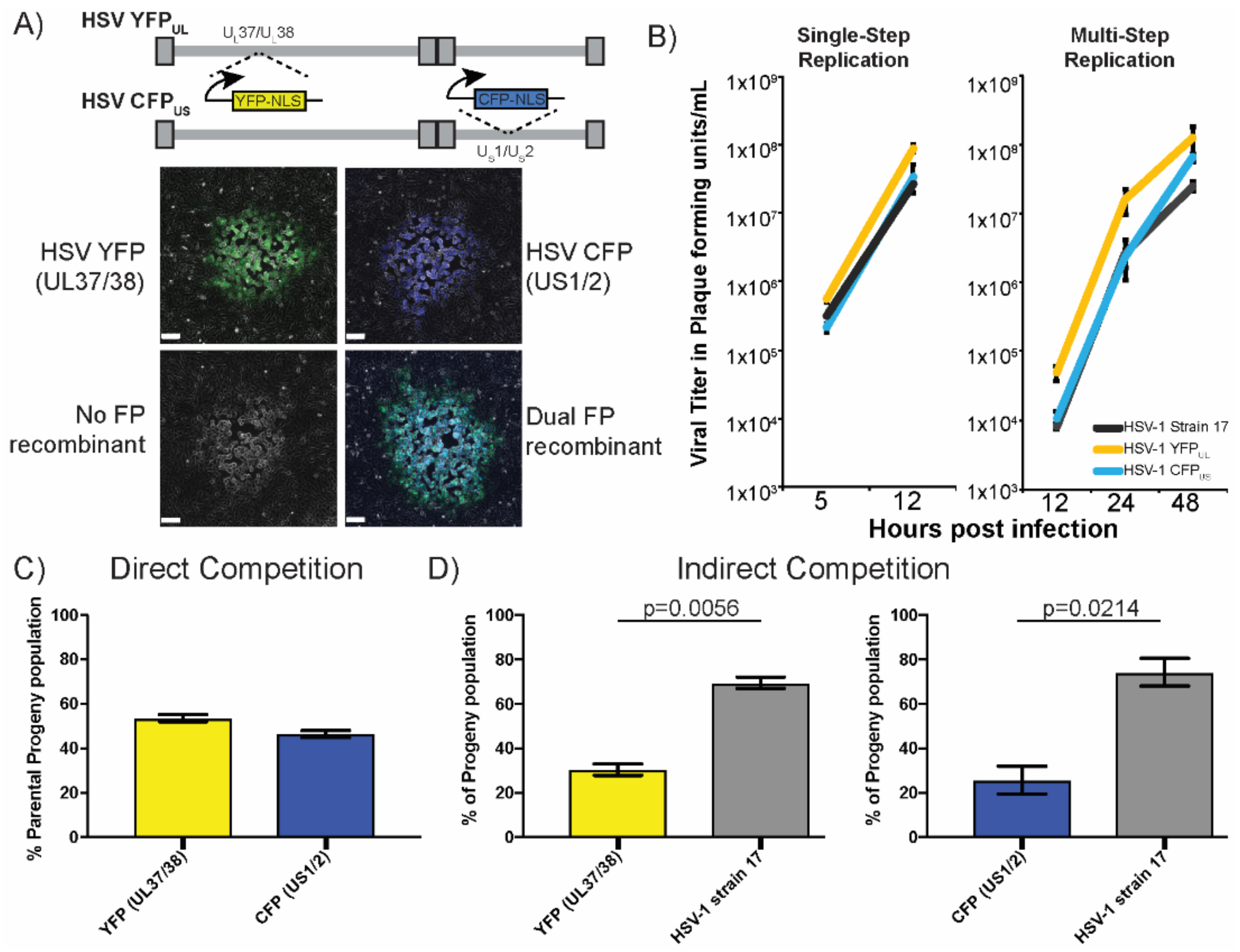
3.1. Adapting Two-Color Fluorescent Protein Expression to Monitor Recombination
3.2. Evaluation of Intergenomic Recombination
3.3. Stable Distribution of Recombinant Progeny during Sequential Passage
3.4. Recombination during Transneuronal Spread
3.5. Recombinant Progeny Production during Transneuronal Spread In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Smith, J.S.; Robinson, N.J. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: A global review. J. Infect. Dis. 2002, 186 (Suppl. 1), S3–S28. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.J.; Roizman, B. Herpes simplex virus infections. Lancet 2001, 357, 1513–1518. [Google Scholar] [CrossRef]
- Granerod, J.; Ambrose, H.E.; Davies, N. Causes of encephalitis and differences in their clinical presentations in England: A multicentre, population-based prospective study. Lancet 2010, 10, 835–844. [Google Scholar] [CrossRef]
- Tsatsos, M.; MacGregor, C.; Athanasiadis, I.; Moschos, M.M.; Hossain, P.; Anderson, D. Herpes simplex virus keratitis: An update of the pathogenesis and current treatment with oral and topical antiviral agents. Clin. Exp. Ophthalmol. 2016, 44, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Loncoman, C.A.; Vaz, P.K.; Coppo, M.J.; Hartley, C.A.; Morera, F.J.; Browning, G.F.; Devlin, J.M. Natural recombination in alphaherpesviruses: Insights into viral evolution through full genome sequencing and sequence analysis. Infect. Genet. Evol. 2017, 49, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Thiry, E.; Meurens, F.O.; Muylkens, B.T.; McVoy, M.; Gogev, S.; Thiry, J.; Vanderplasschen, A.; Epstein, A.; Keil, G.N.; Schynts, F.D.R. Recombination in alphaherpesviruses. Rev. Med. Virol. 2005, 15, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Burrel, S.; Boutolleau, D.; Ryu, D.; Agut, H.; Merkel, K.; Leendertz, F.H.; Calvignac-Spencer, S. Ancient Recombination Events between Human Herpes Simplex Viruses. Mol. Biol. Evol. 2017, 34, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Szpara, M.L.; Gatherer, D.; Ochoa, A.; Greenbaum, B.; Dolan, A.; Bowden, R.J.; Enquist, L.W.; Legendre, M.; Davison, A.J. Evolution and diversity in human herpes simplex virus genomes. J. Virol. 2014, 88, 1209–1227. [Google Scholar] [CrossRef] [PubMed]
- Kolb, A.W.; Ané, C.; Brandt, C.R. Using HSV-1 Genome Phylogenetics to Track Past Human Migrations. PLoS ONE 2013, 8, e76267. [Google Scholar] [CrossRef] [PubMed]
- Bowden, R.; Sakaoka, H.; Donnelly, P.; Ward, R. High recombination rate in herpes simplex virus type 1 natural populations suggests significant co-infection. Infect. Genet. Evol. 2004, 4, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Koelle, D.M.; Koelle, D.M.; Norberg, P.; Norberg, P.; Fitzgibbon, M.P.; Fitzgibbon, M.P.; Russell, R.M.; Greninger, A.L.; Huang, M.-L.; Stensland, L.; et al. Worldwide circulation of HSV-2 × HSV-1 recombinant strains. Sci. Rep. 2017, 7, 44084. [Google Scholar] [CrossRef] [PubMed]
- Norberg, P.; Depledge, D.P.; Kundu, S.; Atkinson, C.; Brown, J.; Haque, T.; Hussaini, Y.; MacMahon, E.; Molyneaux, P.; Papaevangelou, V.; et al. Recombination of Globally Circulating Varicella Zoster Virus. J. Virol. 2015, 89, 7133–7146. [Google Scholar] [CrossRef] [PubMed]
- Esparza, J.; Benyesh-Melnick, B.; Schaffer, P.A. Intertypic complementation and recombination between temperature-sensitive mutants of herpes simplex virus types 1 and 2. Virology 1976, 70, 372–384. [Google Scholar] [CrossRef]
- Dasgupta, U.B.; Summers, W.C. Genetic recombination of Herpes simplex virus, the role of the host cell and UV-irradiation of the virus. Mol. Genet. Genom. 1980, 178, 617–623. [Google Scholar] [CrossRef]
- Wildy, P. Recombination with Herpes Simplex Virus. Microbiology 1955, 13, 346–360. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.; Weller, S. The Role of DNA Recombination in Herpes Simplex Virus DNA Replication. IUBMB Life 2003, 55, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Dutch, R.E.; Bianchi, V.; Lehman, I.R. Herpes simplex virus type 1 DNA replication is specifically required for high-frequency homologous recombination between repeated sequences. J. Virol. 1995, 69, 3084–3089. [Google Scholar] [PubMed]
- Kolb, A.W.; Lee, K.; Larsen, I.; Craven, M.; Brandt, C.R. Quantitative Trait Locus Based Virulence Determinant Mapping of the HSV-1 Genome in Murine Ocular Infection: Genes Involved in Viral Regulatory and Innate Immune Networks Contribute to Virulence. PLoS Pathog. 2016, 12, e1005499. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kolb, A.W.; Sverchkov, Y.; Cuellar, J.A.; Craven, M.; Brandt, C.R. Recombination Analysis of Herpes Simplex Virus Type 1 Reveals a Bias towards GC Content and the Inverted Repeat Regions. J. Virol. 2015, 89, 7214–7223. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, T.; Arii, J.; Akashi, H.; Kawaguchi, Y. Identification of multiple sites suitable for insertion of foreign genes in herpes simplex virus genomes. Microbiol. Immunol. 2008, 53, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Kobiler, O.; Enquist, L.W. Alphaherpesvirus axon-to-cell spread involves limited virion transmission. Proc. Natl. Acad. Sci. USA 2012, 109, 17046–17051. [Google Scholar] [CrossRef] [PubMed]
- Criddle, A.; Thornburg, T.; Kochetkova, I.; DePartee, M.; Taylor, M.P. gD-Independent Superinfection Exclusion of Alphaherpesviruses. J. Virol. 2016, 90, 4049–4058. [Google Scholar] [CrossRef] [PubMed]
- Ch’ng, T.H.; Enquist, L.W. An in vitro system to study trans-neuronal spread of pseudorabies virus infection. Vet. Microbiol. 2006, 113, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Curanovic, D.; Ch’ng, T.H.; Szpara, M.; Enquist, L. Compartmented neuron cultures for directional infection by alpha herpesviruses. Curr. Protoc. Cell Biol. 2009. [Google Scholar] [CrossRef]
- Card, J.P.; Enquist, L.W. Use and Visualization of Neuroanatomical Viral Transneuronal Tracers. In Visualization Techniques; Humana Press: Totowa, NJ, USA, 2012; pp. 225–268. [Google Scholar]
- Card, J.P.; Enquist, L.W. Transneuronal circuit analysis with pseudorabies viruses. Curr. Protoc. Neurosci. 2001, 68, 1–5. [Google Scholar]
- Rosato, P.C.; Leib, D.A. Neuronal Interferon Signaling Is Required for Protection against Herpes Simplex Virus Replication and Pathogenesis. PLoS Pathog. 2015, 11, e1005028. [Google Scholar] [CrossRef] [PubMed]
- Card, J.P.; Whealy, M.E.; Robbins, A.K.; Moore, R.Y.; Enquist, L.W. Two alpha-herpesvirus strains are transported differentially in the rodent visual system. Neuron 1991, 6, 957–969. [Google Scholar] [CrossRef]
- Pickard, G.E.; Smeraski, C.A.; Tomlinson, C.C.; Banfield, B.W.; Kaufman, J.; Wilcox, C.L.; Enquist, L.W.; Sollars, P.J. Intravitreal injection of the attenuated pseudorabies virus PRV Bartha results in infection of the hamster suprachiasmatic nucleus only by retrograde transsynaptic transport via autonomic circuits. J. Neurosci. 2002, 22, 2701–2710. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C. Genetics of natural resistance to herpesvirus infections in mice. Nature 1975, 258, 152–153. [Google Scholar] [CrossRef] [PubMed]
- Hogue, I.; Bosse, J.; Engel, E.; Scherer, J.; Hu, J.-R.; del Rio, T.; Enquist, L. Fluorescent Protein Approaches in Alpha Herpesvirus Research. Viruses 2015, 7, 5933–5961. [Google Scholar] [CrossRef] [PubMed]
- Dangler, C.A.; Deaver, R.E.; Kolodziej, C.M. Genetic recombination between two strains of Aujeszky’s disease virus at reduced multiplicity of infection. J. Gen. Virol. 1994, 75 Pt 2, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.D.; Elias, P. Recombination during early herpes simplex virus type 1 infection is mediated by cellular proteins. J. Biol. Chem. 2001, 276, 2905–2913. [Google Scholar] [CrossRef] [PubMed]
- Muylkens, B.; Farnir, F.; Meurens, F.; Schynts, F.; Vanderplasschen, A.; Georges, M.; Thiry, E. Coinfection with Two Closely Related Alphaherpesviruses Results in a Highly Diversified Recombination Mosaic Displaying Negative Genetic Interference. J. Virol. 2009, 83, 3127–3137. [Google Scholar] [CrossRef] [PubMed]
- Kobiler, O.; Brodersen, P.; Taylor, M.P.; Ludmir, E.B.; Enquist, L.W. Herpesvirus replication compartments originate with single incoming viral genomes. MBio 2011, 2, e00278-11. [Google Scholar] [CrossRef] [PubMed]
- Kobiler, O.; Lipman, Y.; Therkelsen, K.; Daubechies, I.; Enquist, L.W. Herpesviruses carrying a Brainbow cassette reveal replication and expression of limited numbers of incoming genomes. Nat. Commun. 2010, 1, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Roizman, B.; Jacob, R.J.; Knipe, D.M.; Morse, L.S.; Ruyechan, W.T. On the Structure, Functional Equivalence, and Replication of the Four Arrangements of Herpes Simplex Virus DNA. Cold Spring Harb. Symp. Quant. Biol. 1979, 43, 809–826. [Google Scholar] [CrossRef] [PubMed]
- Glazenburg, K.L.; Moormann, R.J.; Kimman, T.G.; Gielkens, A.L.; Peeters, B.P. In vivo recombination of pseudorabies virus strains in mice. Virus Res. 1994, 34, 115–126. [Google Scholar] [CrossRef]
- Javier, R.T.; Sedarati, F.; Stevens, J.G. Two avirulent herpes simplex viruses generate lethal recombinants in vivo. Science 1986, 234, 746–748. [Google Scholar]
- Herr, A.E.; Hain, K.S.; Taylor, M.P. Limitations on the Multiplicity of Cellular Infection during Human Alphaherpesvirus Disease. Curr. Clin. Microbiol. Rep. 2017, 4, 167–174. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Law, G.A.; Herr, A.E.; Cwick, J.P.; Taylor, M.P. A New Approach to Assessing HSV-1 Recombination during Intercellular Spread. Viruses 2018, 10, 220. https://doi.org/10.3390/v10050220
Law GA, Herr AE, Cwick JP, Taylor MP. A New Approach to Assessing HSV-1 Recombination during Intercellular Spread. Viruses. 2018; 10(5):220. https://doi.org/10.3390/v10050220
Chicago/Turabian StyleLaw, Gabrielle A., Alix E. Herr, James P. Cwick, and Matthew P. Taylor. 2018. "A New Approach to Assessing HSV-1 Recombination during Intercellular Spread" Viruses 10, no. 5: 220. https://doi.org/10.3390/v10050220
APA StyleLaw, G. A., Herr, A. E., Cwick, J. P., & Taylor, M. P. (2018). A New Approach to Assessing HSV-1 Recombination during Intercellular Spread. Viruses, 10(5), 220. https://doi.org/10.3390/v10050220