Within-Host Recombination in the Foot-and-Mouth Disease Virus Genome

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Deep Sequencing
2.2. Multi-Swarm/Quasi-Species Structure
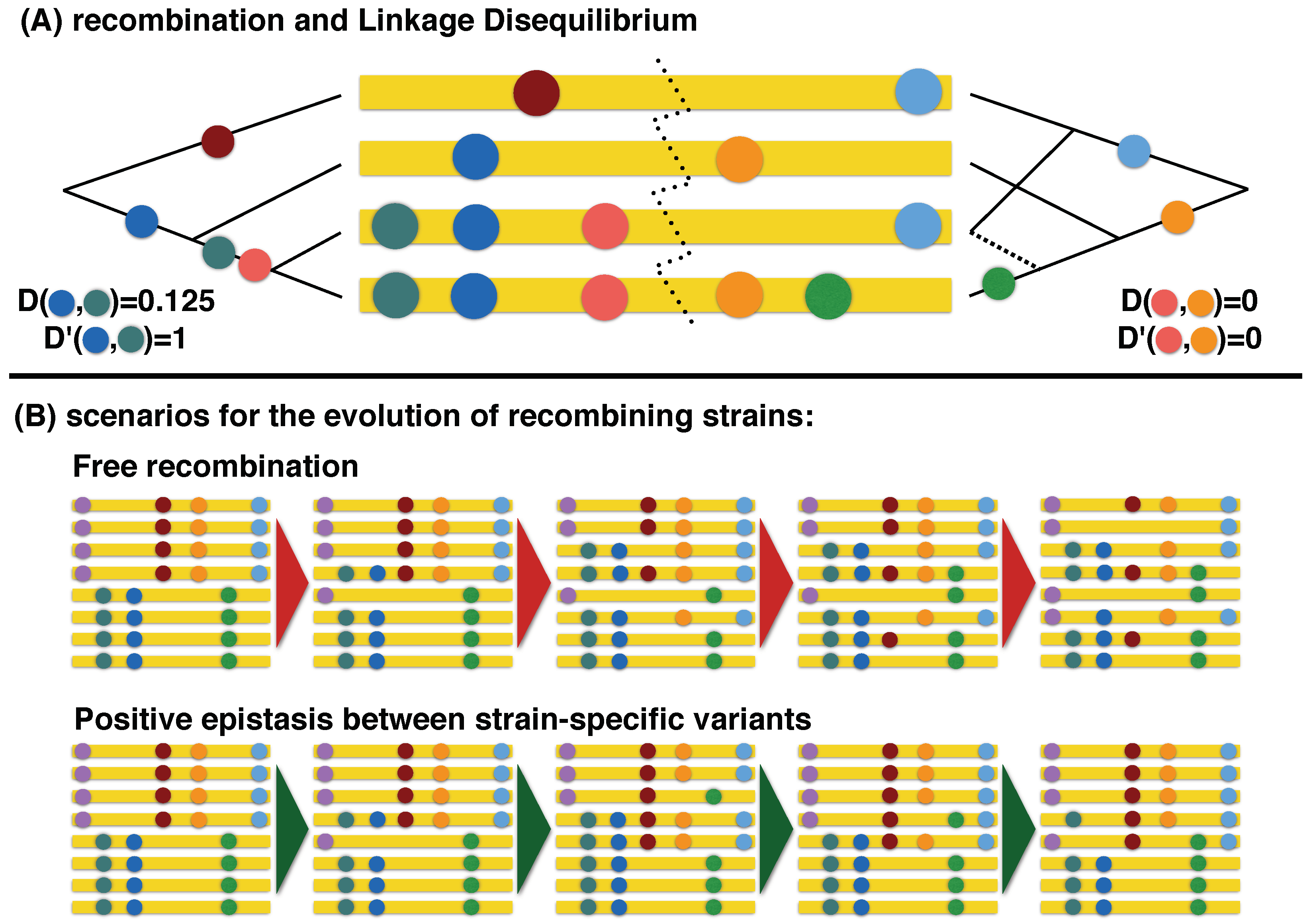
2.3. Inference of Recombination
2.4. Phylogenetic Inference of Recombination Rates
3. Results
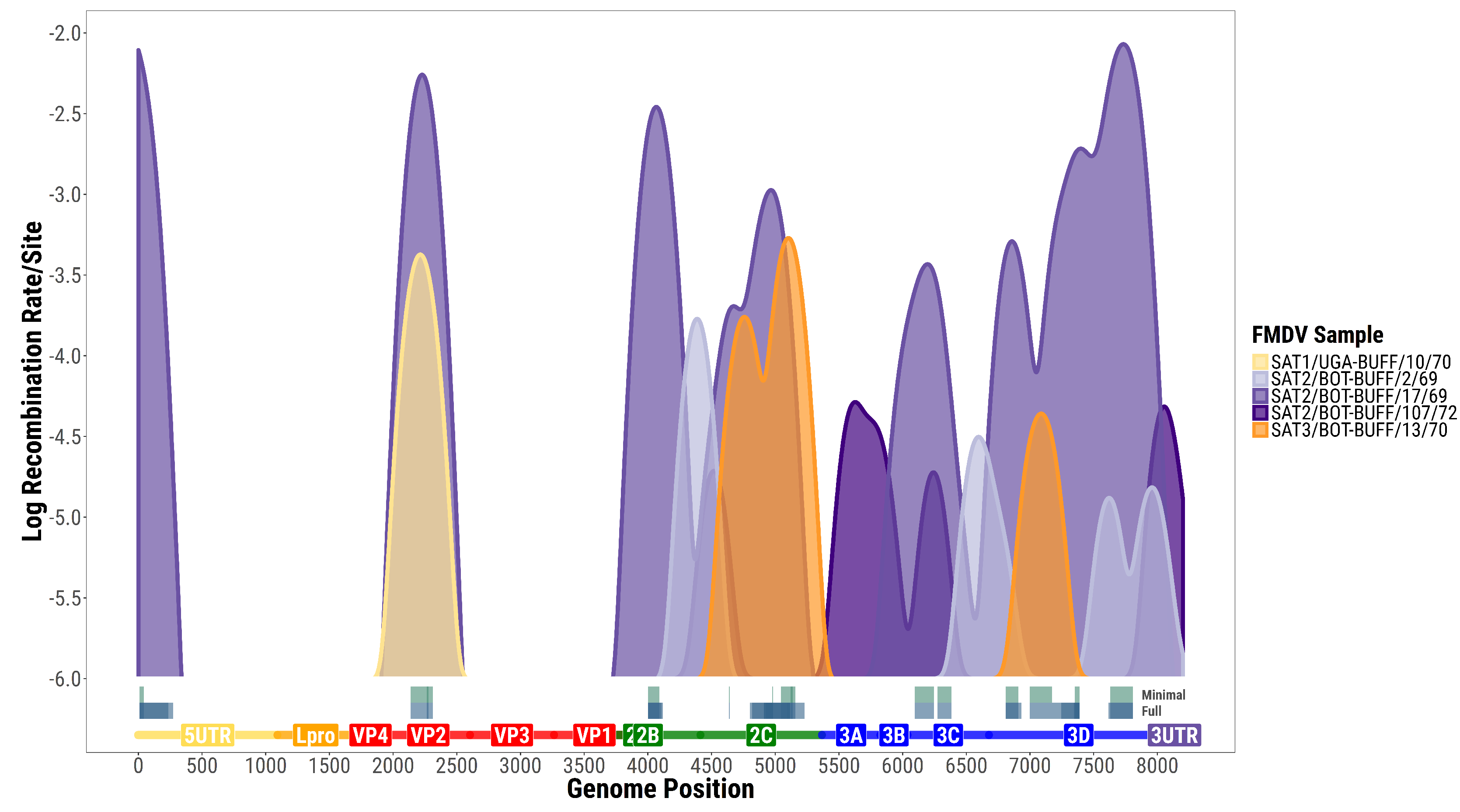
3.1. Recombination Events and Rates
3.2. Within-Host versus Phylogenetic Recombination
4. Discussion
4.1. Impact of Selection against Recombinants
4.2. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Knowles, N.; Hovi, T.; Hyypiä, T.; King, A.; Lindberg, A.M.; Pallansch, M.; Palmenberg, A.; Simmonds, P.; Skern, T.; Stanway, G.; et al. Picornaviridae . In Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: San Diego, CA, USA, 2012; pp. 855–880. [Google Scholar]
- Alexandersen, S.; Zhang, Z.; Donaldson, A.; Garland, A. The pathogenesis and diagnosis of foot-and-mouth disease. J. Comp. Pathol. 2003, 129, 1–36. [Google Scholar] [CrossRef]
- Mason, P.W.; Grubman, M.J.; Baxt, B. Molecular basis of pathogenesis of FMDV. Virus Res. 2003, 91, 9–32. [Google Scholar] [CrossRef]
- Grubman, M.J.; Baxt, B. Foot-and-mouth disease. Clin. Microbiol. Rev. 2004, 17, 465–493. [Google Scholar] [CrossRef] [PubMed]
- Knowles, N.; Samuel, A. Molecular epidemiology of foot-and-mouth disease virus. Virus Res. 2003, 91, 65–80. [Google Scholar] [CrossRef]
- Paton, D.J.; Sumption, K.J.; Charleston, B. Options for control of foot-and-mouth disease: Knowledge, capability and policy. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2657–2667. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Holland, J. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997, 51, 151–178. [Google Scholar] [CrossRef] [PubMed]
- King, A.M. Genetic recombination in positive strand RNA viruses. In RNA Genetics, Volume II, Retroviruses, Viroids, and RNA Recombination; CRC Press: Albany, NY, USA, 1988; pp. 149–165. [Google Scholar]
- Carrillo, C.; Tulman, E.; Delhon, G.; Lu, Z.; Carreno, A.; Vagnozzi, A.; Kutish, G.; Rock, D. Comparative genomics of foot-and-mouth disease virus. J. Virol. 2005, 79, 6487–6504. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Rogers, N.; McClellan, D.A.; Crandall, K.A. The evolution of foot-and-mouth disease virus: Impacts of recombination and selection. Infect. Genet. Evolut. 2008, 8, 786–798. [Google Scholar] [CrossRef] [PubMed]
- McCahon, D.; King, A.M.; Roe, D.S.; Slade, W.R.; Newman, J.W.; Cleary, A.M. Isolation and biochemical characterization of intertypic recombinants of foot-and-mouth disease virus. Virus Res. 1985, 3, 87–100. [Google Scholar] [CrossRef]
- Lee, K.N.; Oem, J.K.; Park, J.H.; Kim, S.M.; Lee, S.Y.; Tserendorj, S.; Sodnomdarjaa, R.; Joo, Y.S.; Kim, H. Evidence of recombination in a new isolate of foot-and-mouth disease virus serotype Asia 1. Virus Res. 2009, 139, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Balinda, S.N.; Siegismund, H.R.; Muwanika, V.B.; Sangula, A.K.; Masembe, C.; Ayebazibwe, C.; Normann, P.; Belsham, G.J. Phylogenetic analyses of the polyprotein coding sequences of serotype O foot-and-mouth disease viruses in East Africa: Evidence for interserotypic recombination. Virol. J. 2010, 7, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamal, S.M.; Ferrari, G.; Ahmed, S.; Normann, P.; Belsham, G.J. Molecular characterization of serotype Asia-1 foot-and-mouth disease viruses in Pakistan and Afghanistan; emergence of a new genetic Group and evidence for a novel recombinant virus. Infect. Genet. Evolut. 2011, 11, 2049–2062. [Google Scholar] [CrossRef] [PubMed]
- Haydon, D.T.; Bastos, A.D.; Awadalla, P. Low linkage disequilibrium indicative of recombination in foot-and-mouth disease virus gene sequence alignments. J. Gen. Virol. 2004, 85, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.; O’neill, H.; Maree, F.; Blignaut, B.; Carrillo, C.; Rodriguez, L.; Haydon, D. Mosaic structure of foot-and-mouth disease virus genomes. J. Gen. Virol. 2007, 88, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Tosh, C.; Hemadri, D.; Sanyal, A. Evidence of recombination in the capsid-coding region of type A foot-and-mouth disease virus. J. Gen. Virol. 2002, 83, 2455–2460. [Google Scholar] [CrossRef] [PubMed]
- Tosh, C.; Sanyal, A.; Hemadri, D. Genetic and antigenic analysis of a recombinant foot-and-mouth disease virus. Curr. Sci. 2002, 1016–1019. [Google Scholar]
- Heath, L.; Van Der Walt, E.; Varsani, A.; Martin, D.P. Recombination patterns in aphthoviruses mirror those found in other picornaviruses. J. Virol. 2006, 80, 11827–11832. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, L.; Perez, E.; Zhang, F.; Maree, F.; Charleston, B.; Ribeca, P. Within-Host Recombination in Structural Proteins of the Foot-and-Mouth Disease Virus. bioRxiv. 2018. Available online: https://www.biorxiv.org/content/early/2018/02/24/271239.full.pdf (accessed on 24 April 2018).
- Logan, G. The Molecular and Genetic Evolution of Foot-and-Mouth Disease Virus. Ph.D. Thesis, University of Glasgow, Glasgow, UK, 2017. Available online: http://theses.gla.ac.uk/7877/1/2017LoganPhD.pdf (accessed on 24 April 2018).
- Ramirez-Carvajal, L.; Pauszek, S.J.; Ahmed, Z.; Farooq, U.; Naeem, K.; Shabman, R.S.; Stockwell, T.B.; Rodriguez, L.L. Genetic stability of foot-and-mouth disease virus during long-term infections in natural hosts. PLoS ONE 2018, 13, e0190977. [Google Scholar] [CrossRef] [PubMed]
- Lasecka-Dykes, L.; Wright, C.F.; Di Nardo, A.; Logan, G.; Mioulet, V.; Jackson, T.; Tuthill, T.J.; Knowles, N.J.; King, D.P. Full Genome Sequencing Reveals New Southern African Territories Genotypes Bringing Us Closer to Understanding True Variability of Foot-and-Mouth Disease Virus in Africa. Viruses 2018, 10. [Google Scholar] [CrossRef]
- Logan, G.; Freimanis, G.L.; King, D.J.; Valdazo-González, B.; Bachanek-Bankowska, K.; Sanderson, N.D.; Knowles, N.J.; King, D.P.; Cottam, E.M. A universal protocol to generate consensus level genome sequences for foot-and-mouth disease virus and other positive-sense polyadenylated RNA viruses using the Illumina MiSeq. BMC Genom. 2014, 15, 828. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, A.; Andino, R. Library preparation for highly accurate population sequencing of RNA viruses. Nat. Protoc. 2014, 9, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Marco-Sola, S.; Sammeth, M.; Guigó, R.; Ribeca, P. The GEM mapper: Fast, accurate and versatile alignment by filtration. Nat. Methods 2012, 9, 1185. [Google Scholar] [CrossRef] [PubMed]
- Raineri, E.; Ferretti, L.; Esteve-Codina, A.; Nevado, B.; Heath, S.; Pérez-Enciso, M. SNP calling by sequencing pooled samples. BMC Bioinform. 2012, 13, 239. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.L.; Clark, A.G.; Clark, A.G. Principles of Population Genetics; Sinauer Associates: Sunderland, MA, USA, 1997. [Google Scholar]
- Hudson, R.R.; Kaplan, N.L. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 1985, 111, 147–164. [Google Scholar] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Franklin, I.; Lewontin, R. Is the gene the unit of selection? Genetics 1970, 65, 707–734. [Google Scholar] [PubMed]
- Wolf, J.B.; Brodie, E.D.; Wade, M.J. Epistasis and the Evolutionary Process; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Jegouic, S.; Joffret, M.L.; Blanchard, C.; Riquet, F.B.; Perret, C.; Pelletier, I.; Colbere-Garapin, F.; Rakoto-Andrianarivelo, M.; Delpeyroux, F. Recombination between polioviruses and co-circulating Coxsackie A viruses: Role in the emergence of pathogenic vaccine-derived polioviruses. PLoS Pathog. 2009, 5, e1000412. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, K.; Baltimore, D. The mechanism of RNA recombination in poliovirus. Cell 1986, 47, 433–443. [Google Scholar] [CrossRef]
- Runckel, C.; Westesson, O.; Andino, R.; DeRisi, J.L. Identification and manipulation of the molecular determinants influencing poliovirus recombination. PLoS Pathog. 2013, 9, e1003164. [Google Scholar] [CrossRef] [PubMed]
- Lowry, K.; Woodman, A.; Cook, J.; Evans, D.J. Recombination in enteroviruses is a biphasic replicative process involving the generation of greater-than genome length ‘imprecise’ intermediates. PLoS Pathog. 2014, 10, e1004191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muslin, C.; Joffret, M.L.; Pelletier, I.; Blondel, B.; Delpeyroux, F. Evolution and emergence of enteroviruses through intra-and inter-species recombination: Plasticity and phenotypic impact of modular genetic exchanges in the 5’untranslated region. PLoS Pathog. 2015, 11, e1005266. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Dolan, P.T.; Goldstein, E.F.; Li, M.; Farkov, M.; Brodsky, L.; Andino, R. Poliovirus intrahost evolution is required to overcome tissue-specific innate immune responses. Nat. Commun. 2017, 8, 375. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Serotype | Host | Avg Read Len | Avg Cov | # SNPs | Avg Freq |
|---|---|---|---|---|---|---|
| SAT3/BOT-BUFF/13/70 | SAT3 | African buffalo | 122 | 10,970 | 58 | 0.36 |
| SAT1/UGA-BUFF/10/70 | SAT1 | African buffalo | 120 | 7843 | 36 | 0.17 |
| SAT2/BOT-BUFF/2/69 | SAT2 | African buffalo | 114 | 9330 | 67 | 0.34 |
| SAT2/BOT-BUFF/17/69 | SAT2 | African buffalo | 119 | 9255 | 115 | 0.19 |
| SAT2/BOT-BUFF/107/72 | SAT2 | African buffalo | 128 | 4735 | 108 | 0.15 |
| SAT1/TAN/22/2012 | SAT1 | cattle | 160 | 11,639 | 31 | 0.17 |
| SAT3/ZAM/P2/96 (NAN-11) | SAT3 | African buffalo | 165 | 3098 | 144 | 0.31 |
| SAT3/ZIM/P6/83 BUFF-16 | SAT3 | African buffalo | 100 | 1413 | 34 | 0.38 |
| Samples from Ramirez et al. [24] | A, O, Asia1 | Asian buffalo | see [24] | see [24] | see [24] | see [24] |
| Sample | Recombination Rate | (“Background”) |
|---|---|---|
| SAT3/BOT-BUFF/13/70 | 0.1 | (0.008) |
| SAT1/UGA-BUFF/10/70 | 0.06 | (0) |
| SAT2/BOT-BUFF/2/69 | 0.011 | (0.002) |
| SAT2/BOT-BUFF/17/69 | 1 | (0.005) |
| SAT2/BOT-BUFF/107/72 | 0.018 | (0.001) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferretti, L.; Di Nardo, A.; Singer, B.; Lasecka-Dykes, L.; Logan, G.; Wright, C.F.; Pérez-Martín, E.; King, D.P.; Tuthill, T.J.; Ribeca, P. Within-Host Recombination in the Foot-and-Mouth Disease Virus Genome. Viruses 2018, 10, 221. https://doi.org/10.3390/v10050221
Ferretti L, Di Nardo A, Singer B, Lasecka-Dykes L, Logan G, Wright CF, Pérez-Martín E, King DP, Tuthill TJ, Ribeca P. Within-Host Recombination in the Foot-and-Mouth Disease Virus Genome. Viruses. 2018; 10(5):221. https://doi.org/10.3390/v10050221
Chicago/Turabian StyleFerretti, Luca, Antonello Di Nardo, Benjamin Singer, Lidia Lasecka-Dykes, Grace Logan, Caroline F. Wright, Eva Pérez-Martín, Donald P. King, Tobias J. Tuthill, and Paolo Ribeca. 2018. "Within-Host Recombination in the Foot-and-Mouth Disease Virus Genome" Viruses 10, no. 5: 221. https://doi.org/10.3390/v10050221
APA StyleFerretti, L., Di Nardo, A., Singer, B., Lasecka-Dykes, L., Logan, G., Wright, C. F., Pérez-Martín, E., King, D. P., Tuthill, T. J., & Ribeca, P. (2018). Within-Host Recombination in the Foot-and-Mouth Disease Virus Genome. Viruses, 10(5), 221. https://doi.org/10.3390/v10050221