1. Introduction
The
Alphaherpesvirinae are a subfamily of the
Herpesviridae that include the human pathogens herpes simplex virus (HSV)-1 and HSV-2. The
UL21 gene encodes a multifunctional protein that is conserved amongst the alphaherpesviruses. The UL21 protein (pUL21) is a component of the tegument, a compartment of the herpesvirus virion located between the capsid and the envelope. Tegument components are delivered to the cell cytoplasm immediately upon viral entry, where they have the opportunity to promote viral infection prior to de novo viral gene expression. In the case of both HSV-1 and HSV-2, pUL21 plays an as yet undefined role early in infection that promotes the initiation of viral gene expression [
1,
2]. As HSV pUL21 can associate with both capsids [
3] and microtubules [
4], it has been proposed that pUL21 may directly facilitate the transport of incoming capsids along microtubules to the nucleus, and that delayed capsid transport in the absence of pUL21 results in the delay of viral gene expression [
1].
HSV pUL21 is also known to play multiple roles late in infection. In HSV-2, pUL21 is required for efficient translocation of capsids from the nucleus to the cytoplasm, where final virion envelopment and maturation takes place. Herpesvirus capsids are assembled within the nucleus, and are too large to exit the nucleus through nuclear pores. Instead, capsids containing viral genomic DNA undergo primary envelopment at the inner nuclear membrane (INM) followed by de-envelopment at the outer nuclear membrane (ONM), to relocate from the nucleus to the cytoplasm. In cells infected with HSV-2 lacking pUL21, capsids accumulated in the nucleoplasm and were rarely observed in the cytoplasm or in the perinuclear space between the INM and the ONM, consistent with a role for pUL21 in nuclear egress [
2]. Accumulation of capsids in the nucleoplasm was not observed in a recent analysis of cells infected with HSV-1 lacking pUL21, however, an increase in the incidence of empty capsids (lacking viral genomic DNA) in both the nucleoplasm and the cytoplasm was noted [
5]. Finally, roles for HSV-1 pUL21 have been described in events occurring later in the infectious cycle. In the cytoplasm, pUL21 forms a complex with the tegument proteins pUL16 and pUL11 on the cytoplasmic tail of the viral glycoprotein, gE [
6,
7,
8]. This complex is required for efficient glycosylation of gE and its trafficking to the cell surface [
7]. Furthermore, deletion of
UL21 from HSV-1 results in the failure of nascent virions to incorporate pUL16 into the tegument [
3]. Crystal structures of both the amino and carboxy termini of HSV-1 pUL21 have been determined [
9,
10] however, both structures are unique, and thus, have not provided further insight into the multiple roles attributed to pUL21 during HSV infection. Intriguingly, during the isolation of the C-terminal domain of pUL21 for crystallization studies, it was noted that this domain co-purified with bacterial RNA, suggesting that pUL21 may have RNA binding capacity [
10].
As pUL21 plays multiple roles in HSV infection, it is not surprising that pUL21 is required for optimal viral replication. What is surprising is that the strictness of this requirement differs between HSV-1 and HSV-2, yet the pUL21s of these distinct virus species are roughly 84% identical. When either homologous recombination or
en passant mutagenesis of a bacterial artificial chromosome (BAC) was used to introduce a deletion into
UL21 of HSV-1 strains F or KOS, the resulting viruses were able to replicate in the absence of complementation, but had growth deficiencies in non-complementing cells, ranging from 3- to 100-fold, and also produced smaller plaques in non-complementing cells [
1,
5,
11]. By contrast, when BAC
en passant mutagenesis was used to introduce a deletion into
UL21 of HSV-2 strain 186, the resulting virus was extremely crippled, due primarily to its nuclear egress defect, and required complementation to replicate [
2]. Thus, there are discrepancies regarding the necessity of pUL21 in HSV replication: pUL21 is essential in the case of HSV-2 and non-essential in the case of HSV-1. What might explain these discrepancies? One possibility is that the results obtained with the single strain of HSV-2 studied by Le Sage et al., strain 186, were not representative of the HSV-2 species as a whole. Another possibility is that pUL21 has species-specific functions during the replication of HSV-2 and HSV-1.
The goals of this study were to establish whether the phenotype of other HSV-2 strains carrying a deletion in UL21 resembled strain 186, and to re-examine whether phenotypic differences exist between HSV-1 and HSV-2. To achieve these goals, CRISPR/Cas9 mutagenesis was used to engineer a panel of UL21 deletion mutants in multiple HSV strains. Comparative analysis of cell-to-cell spread and replication kinetics of this panel of UL21 deletion mutants in non-complementing cells revealed both strain-specific and species-specific differences in the requirement for pUL21 in HSV replication.
2. Materials and Methods
2.1. Cells and Viruses
African green monkey kidney cells (Vero) and murine L cell fibroblasts were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in a 5% CO
2 environment. L cells, 293T cells, and HaCaT cells that stably produce pUL21 of HSV-2 strain 186 (referred to herein as L21, 293T21 and HaCaT21), were similarly maintained. 293T21 and HaCaT21 cells were isolated by retroviral transduction using an amphotropic Phoenix-Moloney murine leukemia virus system, as described previously for the isolation of L21 cells [
2]. All viruses used in this study were propagated in either L21 or HaCaT21 cells. HSV-2 strains SD90e, HG52, and 186 were acquired from Dr. D. M. Knipe (Harvard University), Dr. D. J. McGeoch (MRC Virology Unit, University of Glasgow), and Dr. Y. Kawaguchi (University of Tokyo), respectively; HSV-1 strains KOS and F were acquired from Dr. L. W. Enquist (Princeton University). Infection times, reported in hours post infection (hpi) or days post infection (dpi), refer to the length of time following a one hour inoculation period.
2.2. Construction of UL21 Guide RNA Expression Plasmids
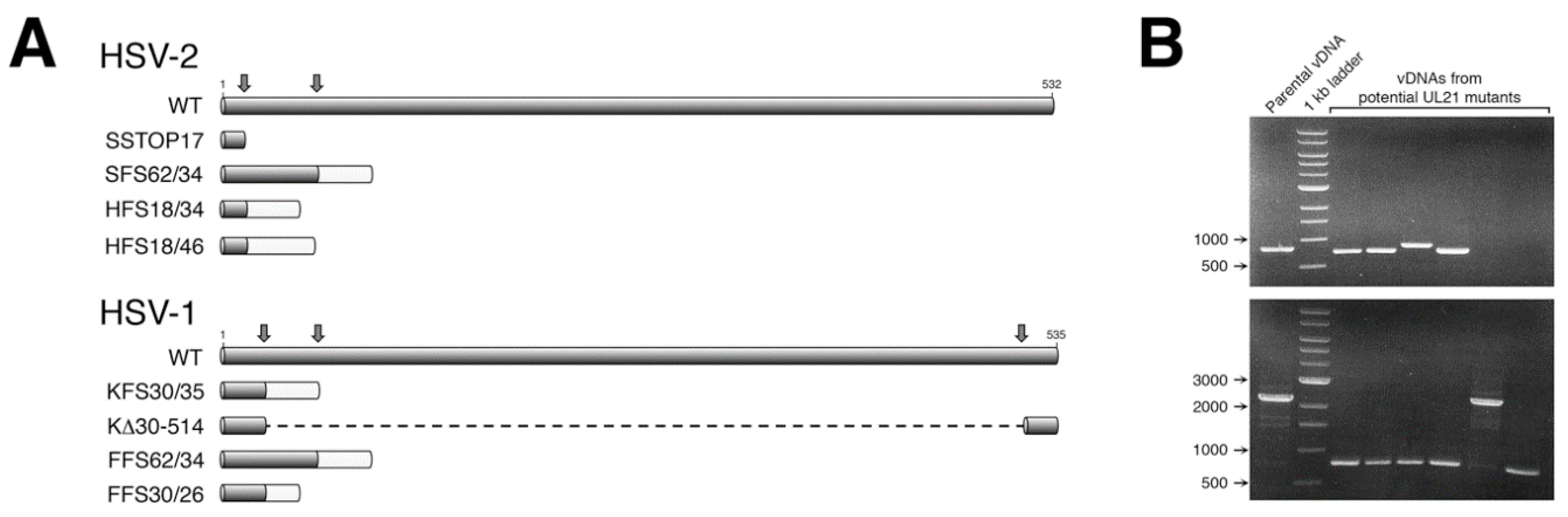
Guide RNAs (gRNAs), used for CRISPR/Cas9 mutagenesis, were expressed from plasmid pX330-U6_Chimeric_BB-CBh-hSpCas9, a gift from Feng Zhang, the Broad Institute of MIT, (Addgene plasmid 42230 [
12]). To construct
UL21-specific gRNA expression plasmids, the top-strand oligonucleotide was annealed to the corresponding bottom-strand oligonucleotide (
Table 1), and the double-stranded DNA product was ligated into pX330-U6_Chimeric_BB-CBh-hSpCas9 plasmid that had been digested with
BbsI. Two gRNAs were designed and utilized in our initial mutagenesis of HSV-2
UL21 (
Figure 1A). For our subsequent mutagenesis of HSV-1
UL21, three gRNAs were designed and utilized (
Figure 1A), allowing both small and large deletions to be introduced into the
UL21 locus (
Figure 1B).
2.3. CRISPR/Cas9 Mutagenesis of the UL21 Locus
Viral genomic DNA (vDNA) of each strain (SD90e, HG52, KOS, or F) was purified as described previously [
13]. Subconfluent monolayers of 293T21 cells growing in 100 mm dishes were co-transfected with 16 μg of vDNA, and 1 μg each of two
UL21 gRNA expression plasmids, using a calcium phosphate co-precipitation method [
14]. Twenty-four hours after transfection, the culture medium was replaced with semisolid medium containing 1% methyl cellulose, to allow for plaque formation. Five to six days later, individual plaques were picked and collected in 300 μL of medium. Picked plaque solutions were used to infect monolayers of L21 cells growing in 6-well dishes. Viral DNA was isolated from a portion of the infected L21 monolayers using a modified Hirt lysis protocol [
15], and used as template for PCR. The
UL21 loci from viruses bearing mutations were sequenced in their entirety to determine the precise nature of the mutation introduced.
2.4. Cell-to-Cell Spread Analysis
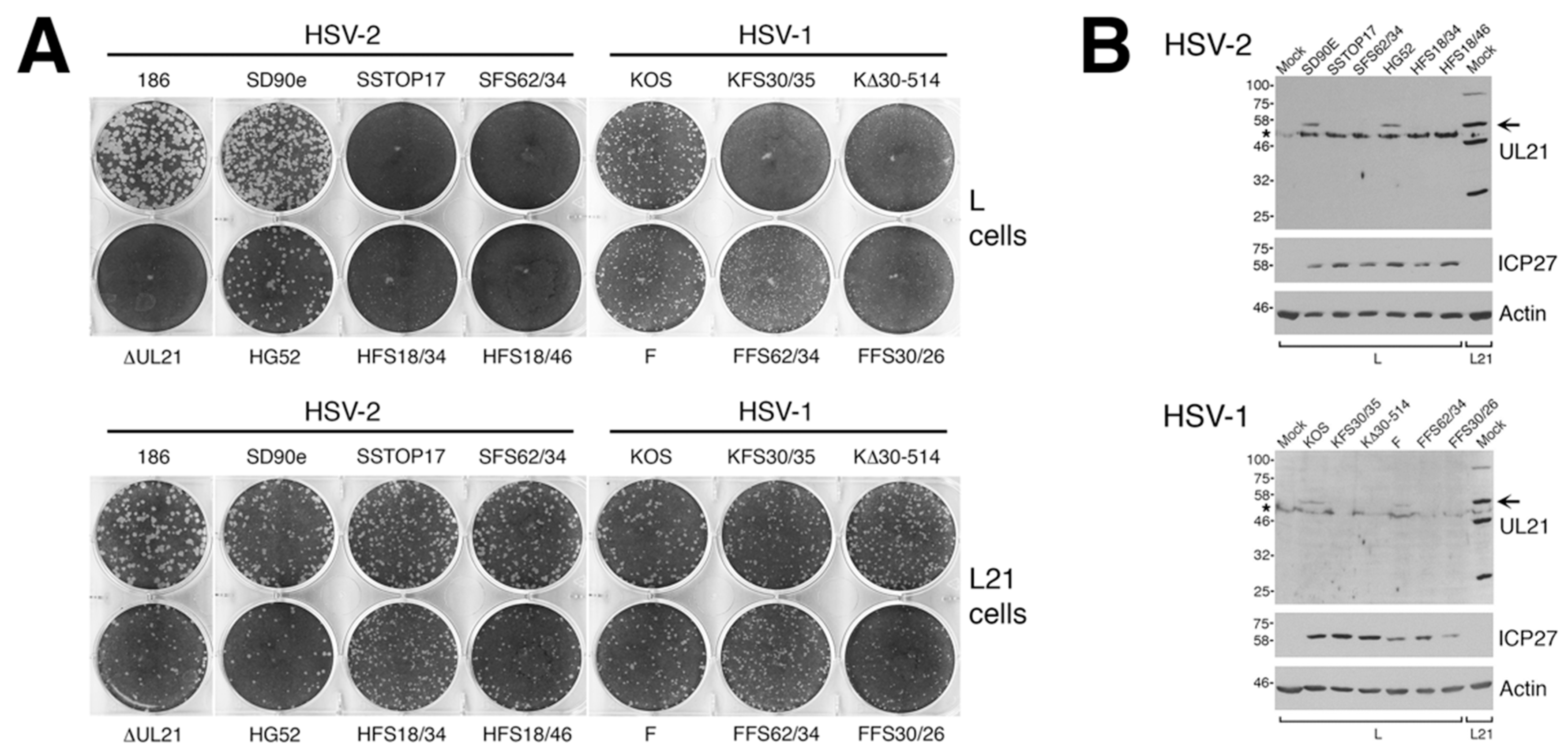
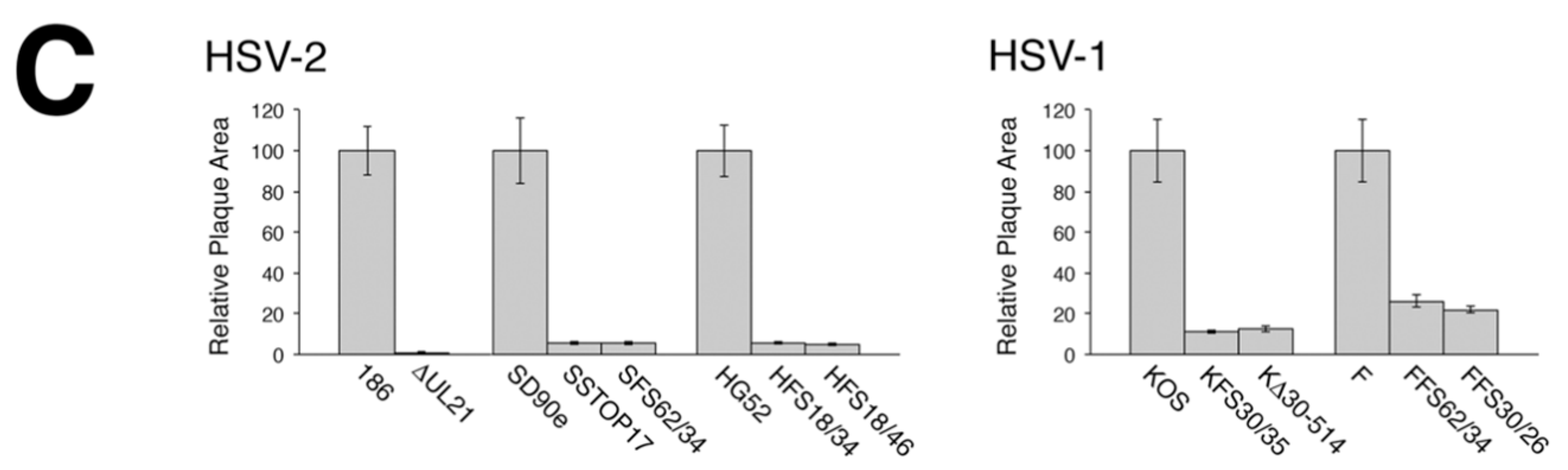
To analyze macroscopic plaque formation, monolayers of Vero cells were infected with equivalent numbers of plaque forming units, overlaid with semisolid medium containing 1% methyl cellulose, then fixed and stained with 70% methanol containing 0.5% methylene blue at 72 hpi. To analyze cell-to-cell spread quantitatively, monolayers of Vero cells growing on glass bottom dishes (MatTek, Ashland, MA, USA) were infected with equivalent numbers of plaque forming units. At 24 hpi, infected monolayers were fixed with 4% paraformaldehyde and stained for the presence of the HSV kinase Us3, as described previously [
16]. Images of plaques were captured on an Olympus FV1000 laser scanning confocal microscope using a 10× objective or on a Nikon TE200 inverted epifluorescence microscope using a 10× objective and a cooled CCD camera. To quantify these results, the numbers of pixels in the area of each plaque were counted using Image-Pro 6.3 software (Media Cybernetics, Bethesda, MD, USA). Results shown were derived from 40 distinct plaques per strain. Single infected cells were not included in this quantification.
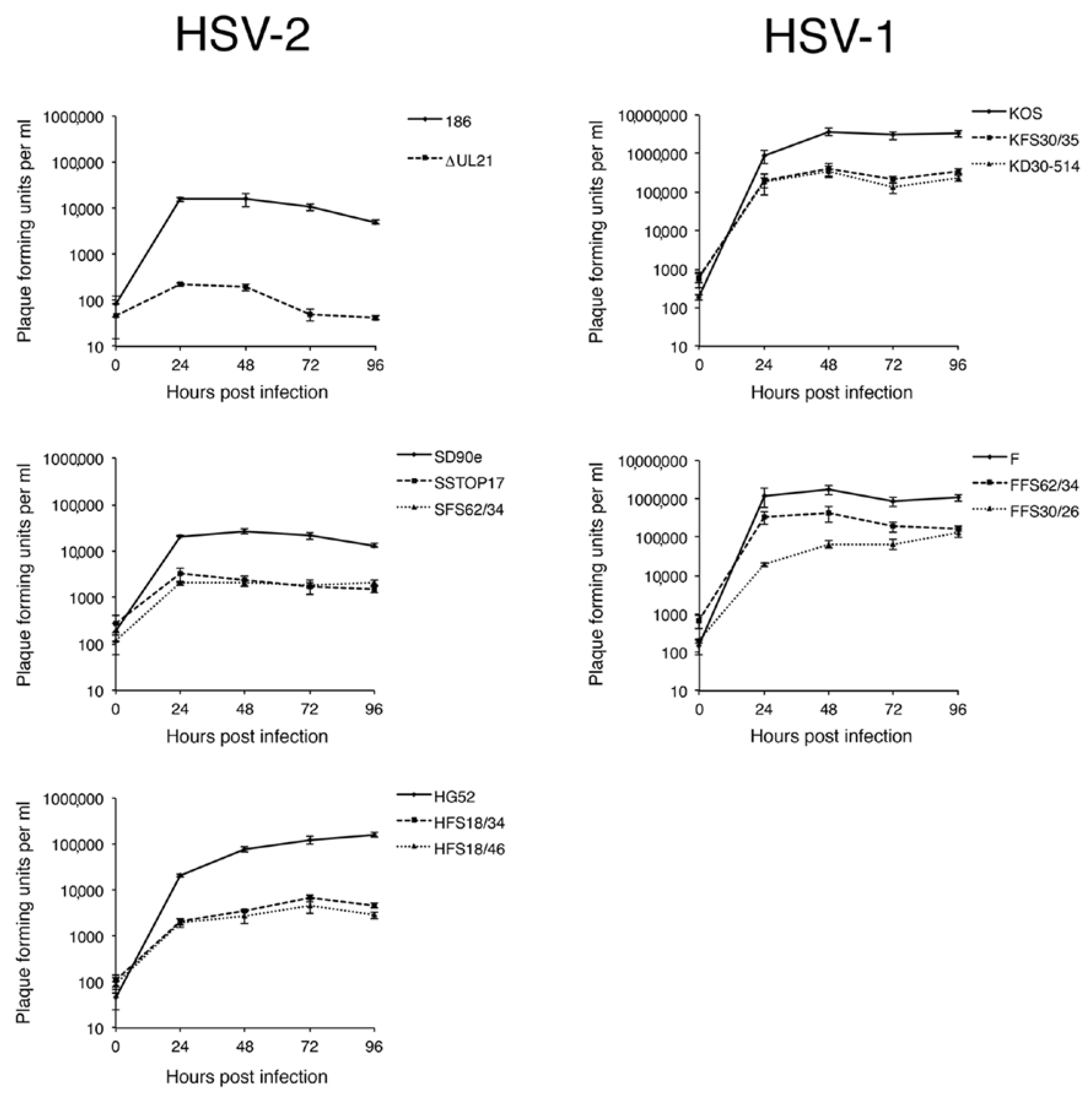
2.5. Virus Replication Analysis
Monolayers of Vero cells in 6-well dishes (approximately 5 × 105 cells per well) were infected at a multiplicity of infection (MOI) of 0.1. All infections were performed in triplicate. After a one hour inoculation period, monolayers were treated for 2 min at 37 °C with low pH citrate buffer (40 mM Na citrate, 10 mM KCl, 0.8% NaCl) to inactivate extracellular virions, washed once with medium, then incubated at 37 °C. Infected monolayers were harvested by scraping cells and medium together at 0, 24, 48, 72, and 96 hpi and stored at −80 °C. Samples were subjected to two freeze/thaw cycles, sonicated briefly in a chilled cup-horn sonicator, and cellular debris removed from the lysate by brief centrifugation at 3000 rpm in a microcentrifuge. The virus titers of the clarified lysates were determined in duplicate by plaque assay on L21 cells.
2.6. Immunological Reagents
Rat polyclonal antiserum against HSV-2 Us3 [
16] was used for indirect immunofluorescence microscopy at a dilution of 1:500; rat polyclonal antiserum against HSV-2 pUL21 [
2] was used for Western blotting at a dilution of 1:600; mouse monoclonal antibody against HSV ICP27 (Virusys, Taneytown, MD, USA) was used for Western blotting at a dilution of 1:500; mouse monoclonal antibody against β-actin (Sigma, St. Louis, MO, USA) was used for Western blotting at a dilution of 1:2000; Alexa Fluor 488 conjugated donkey anti-rat IgG, (Molecular Probes, Eugene, OR, USA) was used for indirect immunofluorescence at a dilution of 1:500; horseradish peroxidase-conjugated goat anti-mouse IgG (Sigma, St. Louis, MO, USA) was used for Western blotting at a dilution of 1:10,000; horseradish peroxidase-conjugated goat anti-rat IgG (Sigma, St. Louis, MO, USA) was used for Western blotting at a dilution of 1:80,000.
2.7. Preparation and Analysis of Whole Cell Lysates
To prepare whole cell lysates of infected cells for Western blot analyses, cells were washed with cold phosphate buffered saline (PBS), then scraped into cold PBS containing protease inhibitors (Roche) plus 5 mM sodium fluoride (New England Biolabs, Ipswich, MA, USA) and 1 mM sodium orthovanadate (New England Biolabs, Ipswich, MA, USA) to inhibit phosphatases. Harvested cells were transferred to a 1.5 mL microfuge tube containing 3× sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer. The lysate was repeatedly passed through a 28 1/2-gauge needle to reduce viscosity, and then heated at 100 °C for 5 min. For Western blot analysis, 10 to 20 μL of whole cell lysate was electrophoresed through SDS 8% polyacrylamide gels. Proteins were transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA), blocked with 3% bovine serum albumin, then probed with appropriate dilutions of primary antibody followed by appropriate dilutions of horseradish peroxidase conjugated secondary antibody. The membranes were treated with Pierce enhanced chemiluminescence substrate (Thermo Scientific, Rockford, IL, USA) and exposed to film.
4. Discussion
A necessary foundation for understanding the multiple roles played by pUL21 during HSV infection is to definitively establish whether phenotypic differences exist between HSV species or between HSV strains of the same species. The work detailed in this study provides this foundation. By utilizing CRISPR/Cas9 mutagenesis, we were able to construct multiple HSVs lacking pUL21 in an efficient manner, enabling side-by-side phenotypic analyses. These analyses revealed that HSV-2 has a stricter requirement for pUL21 in cell-to-cell spread of virus than does HSV-1. Strain-specific differences in this phenotype were also noted. Cell-to-cell spread of HSV-2 strain 186 was particularly crippled in the absence of pUL21. In particular, we noted an abundance of ∆UL21 infected Vero cells that failed to spread infection to adjacent cells (
Figure 3B, arrow). HSV-2 strain 186 lacking pUL21 also showed a greater deficiency in viral replication in comparison to all other HSV strains lacking pUL21. A previous study from our laboratory demonstrated that HSV-2 strain 186 lacking pUL21 displayed a noticeable defect in nuclear egress [
2]. As the results of this study demonstrate that strain 186 lacking pUL21 is an outlier in both cell-to-cell spread of virus and in viral replication kinetics in comparison to other HSV strains lacking pUL21, it will be of considerable interest to investigate whether strain 186 is also an outlier in terms of reliance on pUL21 for nuclear egress.
Strain-specific differences can provide important clues about the function of a viral protein, as well as mechanistic details about a viral process. Thus, investigating the basis of strain-specific differences is a worthwhile endeavor. In this work, we have demonstrated that CRISPR/Cas9 mutagenesis can be used to resolve strain-specific differences. We have also used the methodologies described, herein, to generate deletion mutants of
UL16 in multiple HSV strains as a means of understanding the role of pUL16 in nuclear egress [
17]. We anticipate that CRISPR/Cas9 mutagenesis will be valuable for generating viruses carrying deletions in multiple viral genes that will further inform our understanding of alphaherpesvirus nuclear egress.
CRISPR/Cas9 mutagenesis provides an efficient and more broadly accessible means of introducing deletions into alphaherpesvirus strains of interest. Homologous recombination methodologies that were traditionally used to construct alphaherpesvirus mutants had mutant strain recovery efficiencies of less than 1% [
15], whereas our CRISPR/Cas9 approach had a mutant recovery efficiency greater than 50% (
Figure 1B). More recently, recombineering approaches utilizing select HSV genomes cloned into BACs have been widely used to accurately and efficiently construct HSV mutants. However, the process of constructing a novel BAC is a laborious undertaking [
2,
18,
19,
20]. CRISPR/Cas9 technology can be readily and rapidly applied to any HSV strain, and should allow observations made in laboratory strains of alphaherpesviruses to be more readily investigated in clinical isolates.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}