Histone Modulation Blocks Treg-Induced Foxp3 Binding to the IL-2 Promoter of Virus-Specific CD8+ T Cells from Feline Immunodeficiency Virus-Infected Cats


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cats and FIV Infection
2.2. CD8+T Cell Co-Culture and CFSE Cell Proliferation Assays
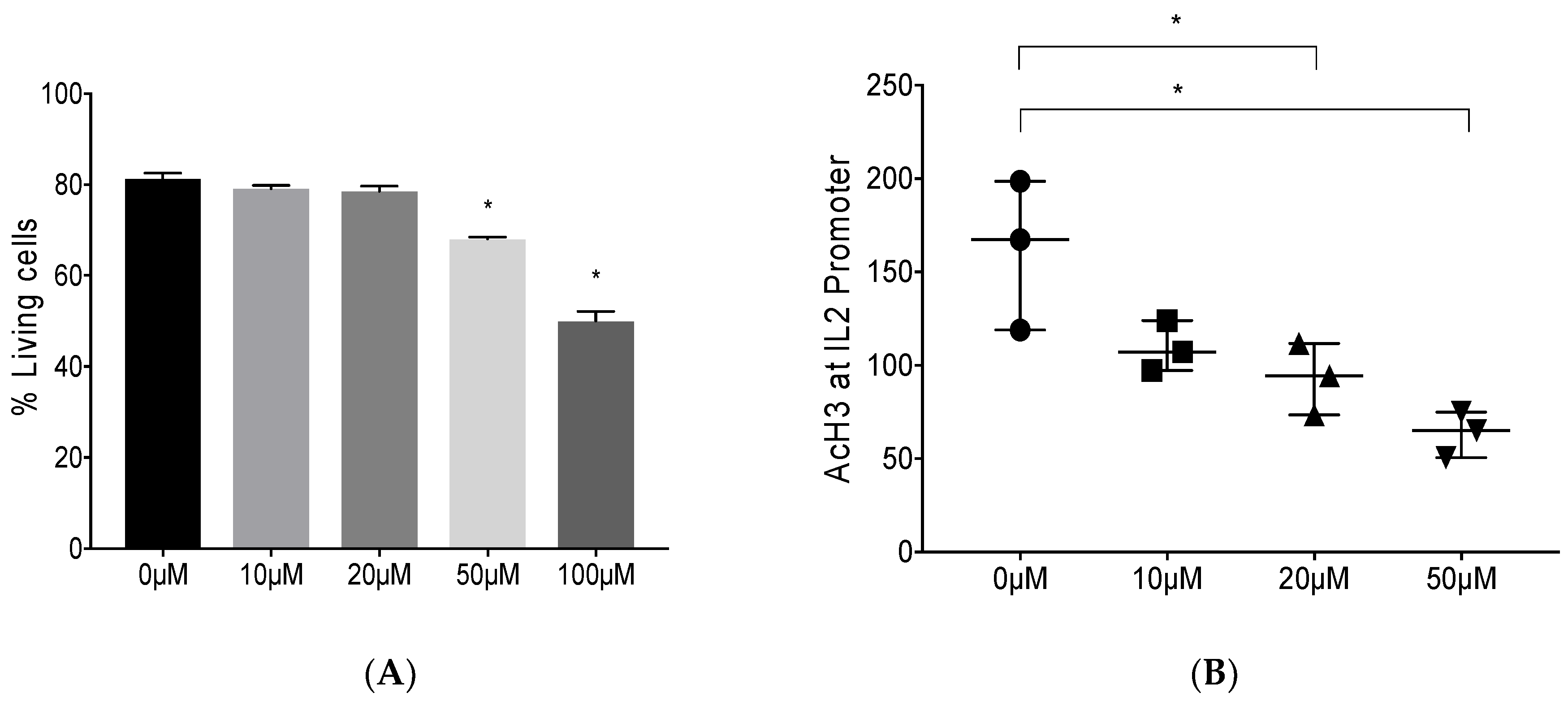
2.3. Mya-1 Cell Culture and Cell Viability
2.4. Chromatin Immunoprecipitation (ChIP) and Acetyl-Histone 3 (AcH3) ChIP Assay
2.5. RNA Extraction, RT and Real-Time PCR Quantification
2.6. Anacardic Acid (AA) Treatment
2.7. Data Analysis
3. Results
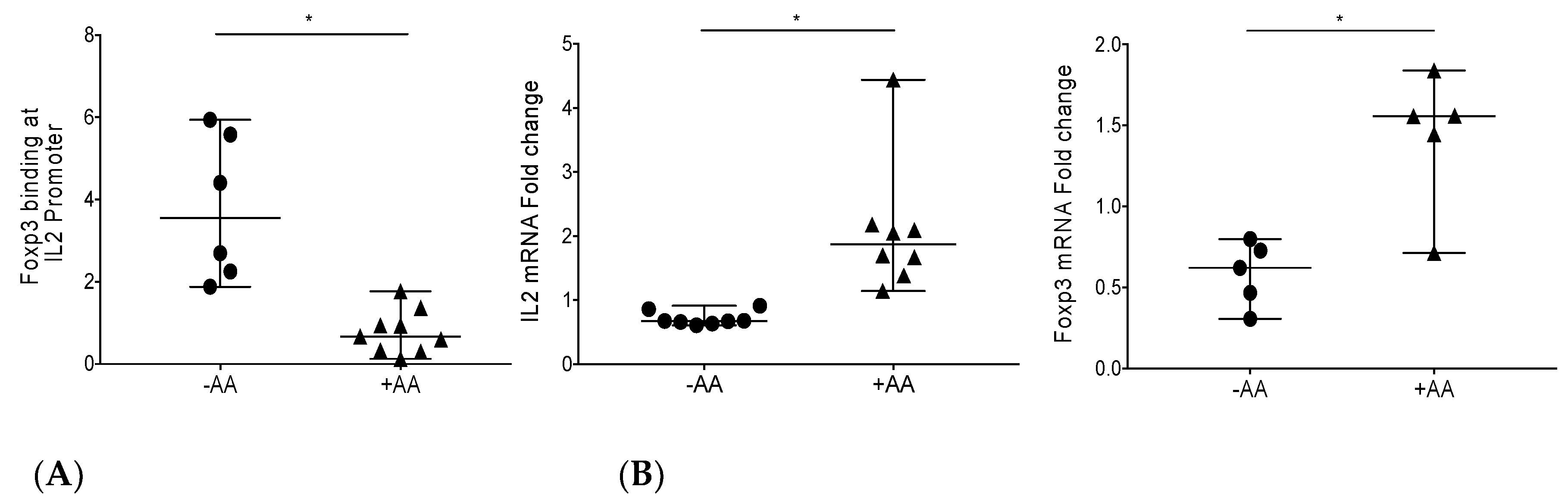
3.1. Epigenetic Modulation by Anacardic Acid (AA) Promotes Histone De-Acetylation and Blocks Foxp3 Binding to the IL-2 Promoter Leading to Higher IL-2 mRNA Levels In Vitro
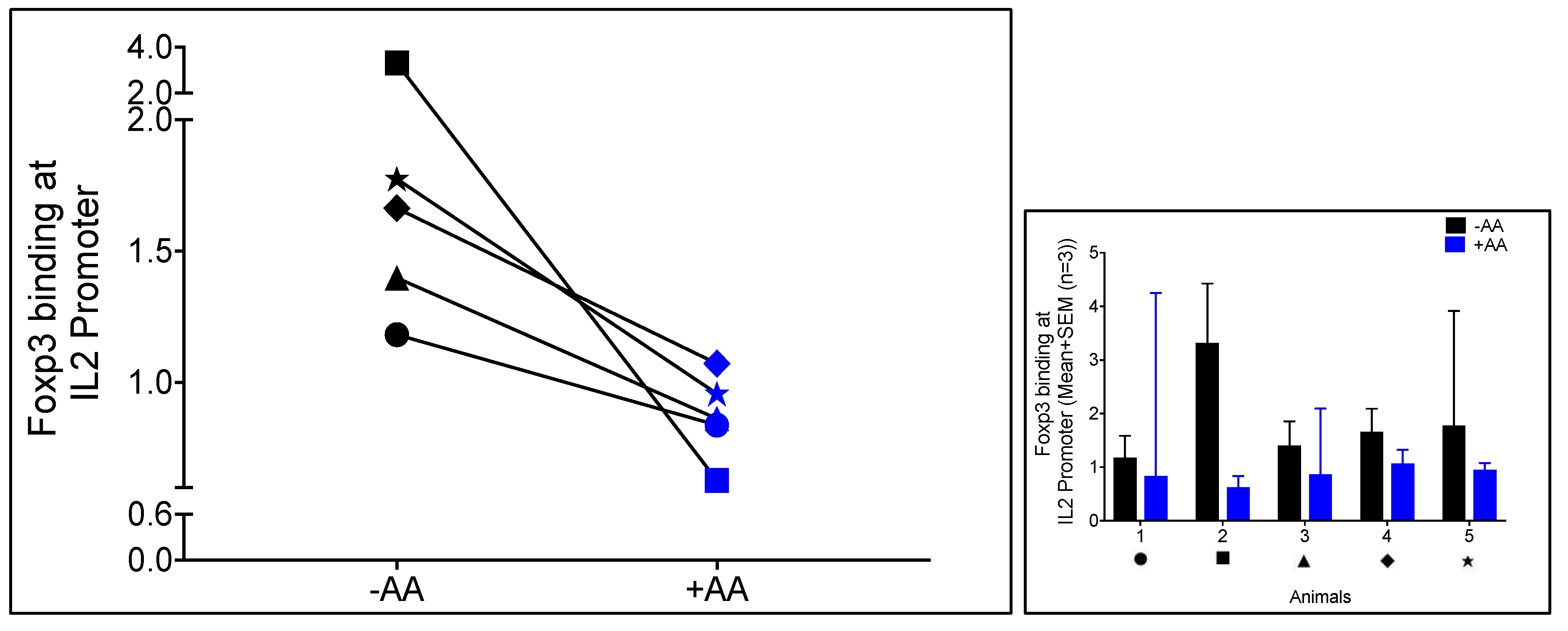
3.2. Anacardic Acid Blocks Foxp3 Binding to the IL-2 Promoter In Virus-Specific CD8+ T Cells Co-Cultured with Autologous Treg Cells in FIV+ Cats
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Appay, V.; Nixon, D.F.; Donahoe, S.M.; Gillespie, G.M.; Dong, T.; King, A.; Ogg, G.S.; Spiegel, H.M.; Conlon, C.; Spina, C.A.; et al. HIV-specific CD8(+) T cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 2000, 192, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Goepfert, P.A.; Bansal, A.; Edwards, B.H.; Ritter, G.D., Jr.; Tellez, I.; McPherson, S.A.; Sabbaj, S.; Mulligan, M.J. A significant number of human immunodeficiency virus epitope-specific cytotoxic T lymphocytes detected by tetramer binding do not produce gamma interferon. J. Virol. 2000, 74, 10249–10255. [Google Scholar] [CrossRef] [PubMed]
- Migueles, S.A.; Connors, M. The Role of CD4(+) and CD8(+) T Cells in Controlling HIV Infection. Curr. Infect. Dis. Rep. 2002, 4, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Obar, J.J.; Lefrancois, L. Early signals during CD8 T cell priming regulate the generation of central memory cells. J. Immunol. 2010, 185, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Kalia, V.; Sarkar, S.; Subramaniam, S.; Haining, W.N.; Smith, K.A.; Ahmed, R. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity 2010, 32, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Pipkin, M.E.; Sacks, J.A.; Cruz-Guilloty, F.; Lichtenheld, M.G.; Bevan, M.J.; Rao, A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 2010, 32, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.J.; Khanolkar, A.; Tebo, A.E.; Zajac, A.J. Maintenance, loss, and resurgence of T cell responses during acute, protracted, and chronic viral infections. J. Immunol. 2004, 172, 4204–4214. [Google Scholar] [CrossRef] [PubMed]
- Zimmerli, S.C.; Harari, A.; Cellerai, C.; Vallelian, F.; Bart, P.A.; Pantaleo, G. HIV-1-specific IFN-gamma/IL-2-secreting CD8 T cells support CD4-independent proliferation of HIV-1-specific CD8 T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 7239–7244. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Deng, K.; Pertea, M.; Rongvaux, A.; Wang, L.; Durand, C.M.; Ghiaur, G.; Lai, J.; McHugh, H.L.; Hao, H.; Zhang, H.; et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature 2015, 517, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Eggena, M.P.; Barugahare, B.; Jones, N.; Okello, M.; Mutalya, S.; Kityo, C.; Mugyenyi, P.; Cao, H. Depletion of regulatory T cells in HIV infection is associated with immune activation. J. Immunol. 2005, 174, 4407–4414. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D.; Jiang, Q.; Zhang, L.; Su, L. Foxp3 and Treg cells in HIV-1 infection and immuno-pathogenesis. Immunol. Res. 2008, 41, 248–266. [Google Scholar] [CrossRef] [PubMed]
- Aandahl, E.M.; Michaelsson, J.; Moretto, W.J.; Hecht, F.M.; Nixon, D.F. Human CD4+ CD25+ regulatory T cells control T-cell responses to human immunodeficiency virus and cytomegalovirus antigens. J. Virol. 2004, 78, 2454–2459. [Google Scholar] [CrossRef] [PubMed]
- Kinter, A.L.; Horak, R.; Sion, M.; Riggin, L.; McNally, J.; Lin, Y.; Jackson, R.; O’Shea, A.; Roby, G.; Kovacs, C.; et al. CD25+ regulatory T cells isolated from HIV-infected individuals suppress the cytolytic and nonlytic antiviral activity of HIV-specific CD8+ T cells in vitro. AIDS Res. Hum. Retrovir. 2007, 23, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Khattri, R.; Cox, T.; Yasayko, S.A.; Ramsdell, F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 2003, 4, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, D.; Tsun, A.; Li, B. FOXP3+ regulatory T cells and their functional regulation. Cell. Mol. Immunol. 2015, 12, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Fogle, J.E.; Mexas, A.M.; Tompkins, W.A.; Tompkins, M.B. CD4(+)CD25(+) T regulatory cells inhibit CD8(+) IFN-gamma production during acute and chronic FIV infection utilizing a membrane TGF-beta-dependent mechanism. AIDS Res. Hum. Retrovir. 2010, 26, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.M.; Akaronu, N.; Thompson, E.M.; Hood, S.F.; Fogle, J.E. Modulating DNA methylation in activated CD8+ T cells inhibits regulatory T cell-induced binding of Foxp3 to the CD8+ T Cell IL-2 promoter. J. Immunol. 2015, 194, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nag, M.; Tuohy, J.L.; De Paris, K.; Fogle, J.E. T Regulatory Cell Induced Foxp3 Binds the IL2, IFNgamma, and TNFalpha Promoters in Virus-Specific CD8(+) T Cells from Feline Immunodeficiency Virus Infected Cats. AIDS Res. Hum. Retrovir. 2017, 34, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Fogle, J.E.; Tompkins, W.A.; Tompkins, M.B. CD4+CD25+ T regulatory cells from FIV+ cats induce a unique anergic profile in CD8+ lymphocyte targets. Retrovirology 2010, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Mexas, A.M.; Fogle, J.E.; Tompkins, W.A.; Tompkins, M.B. CD4+CD25+ regulatory T cells are infected and activated during acute FIV infection. Vet. Immunol. Immunopathol. 2008, 126, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Petty, C.S.; Tompkins, M.B.; Tompkins, W.A. Transforming growth factor-beta/transforming growth factor-betaRII signaling may regulate CD4+CD25+ T-regulatory cell homeostasis and suppressor function in feline AIDS lentivirus infection. J. Acquir. Immune Defic. Syndr. 2008, 47, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.M.; Petty, C.S.; Tompkins, M.B.; Fogle, J.E. CD4+CD25+ T regulatory cells activated during feline immunodeficiency virus infection convert T helper cells into functional suppressors through a membrane-bound TGFbeta/GARP-mediated mechanism. Virol. J. 2014, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Rowell, E.A.; Thomas, R.M.; Hancock, W.W.; Wells, A.D. Transcriptional regulation by Foxp3 is associated with direct promoter occupancy and modulation of histone acetylation. J. Biol. Chem. 2006, 281, 36828–36834. [Google Scholar] [CrossRef] [PubMed]
- Hoji, A.; Coro, A.; Ng, H.L.; Jamieson, B.D.; Yang, O.O. Proliferation and foxp3 expression in virus-specific memory CD8+ T lymphocytes. AIDS Res. Hum. Retrovir. 2008, 24, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Arvey, A.; van der Veeken, J.; Samstein, R.M.; Feng, Y.; Stamatoyannopoulos, J.A.; Rudensky, A.Y. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat. Immunol. 2014, 15, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.K.; Chen, H.M.; Mathis, D.; Benoist, C. Different molecular complexes that mediate transcriptional induction and repression by FoxP3. Nat. Immunol. 2017, 18, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Delcuve, G.P.; Rastegar, M.; Davie, J.R. Epigenetic control. J. Cell. Physiol. 2009, 219, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Yedavalli, V.R.; Jeang, K.T. Methylation: A regulator of HIV-1 replication? Retrovirology 2007, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation, methyltransferases, and cancer. Oncogene 2001, 20, 3139–3155. [Google Scholar] [CrossRef] [PubMed]
- Niller, H.H.; Wolf, H.; Minarovits, J. Epigenetic dysregulation of the host cell genome in Epstein-Barr virus-associated neoplasia. Semin. Cancer Biol. 2009, 19, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Northrop, J.K.; Thomas, R.M.; Wells, A.D.; Shen, H. Epigenetic remodeling of the IL-2 and IFN-gamma loci in memory CD8 T cells is influenced by CD4 T cells. J. Immunol. 2006, 177, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Nakayama-Hosoya, K.; Ishida, T.; Youngblood, B.; Nakamura, H.; Hosoya, N.; Koga, M.; Koibuchi, T.; Iwamoto, A.; Kawana-Tachikawa, A. Epigenetic repression of interleukin 2 expression in senescent CD4+ T cells during chronic HIV type 1 infection. J. Infect. Dis. 2015, 211, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, J.; Woltring, D.; Gerondakis, S.; Shannon, M.F. Histone dynamics on the interleukin-2 gene in response to T-cell activation. Mol. Cell. Biol. 2005, 25, 3209–3219. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kametani, Y.; Katano, I.; Habu, S. T-cell specific enhancement of histone H3 acetylation in 5′ flanking region of the IL-2 gene. Biochem. Biophys. Res. Commun. 2005, 331, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Radde, B.N.; Alizadeh-Rad, N.; Price, S.M.; Schultz, D.J.; Klinge, C.M. Anacardic Acid, Salicylic Acid, and Oleic Acid Differentially Alter Cellular Bioenergetic Function in Breast Cancer Cells. J. Cell. Biochem. 2016, 117, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Hemshekhar, M.; Sebastin Santhosh, M.; Kemparaju, K.; Girish, K.S. Emerging roles of anacardic acid and its derivatives: A pharmacological overview. Basic Clin. Pharmacol. Toxicol. 2012, 110, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Gerhauser, C. Cancer chemoprevention and nutriepigenetics: State of the art and future challenges. Top. Curr. Chem. 2013, 329, 73–132. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Shin, M.H.; Kim, Y.K.; Kim, H.Y.; Lee, Y.R.; Lee, D.H.; Chung, J.H. Anacardic acid ameliorates ultraviolet irradiation-induced damage to human skin. J. Dermatol. Sci. 2017, 86, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, M.B.; Gebhard, D.H.; Bingham, H.R.; Hamilton, M.J.; Davis, W.C.; Tompkins, W.A. Characterization of monoclonal antibodies to feline T lymphocytes and their use in the analysis of lymphocyte tissue distribution in the cat. Vet. Immunol. Immunopathol. 1990, 26, 305–317. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, X.; Chen, S.; Price, B.D. Inhibition of histone acetyltransferase activity by anacardic acid sensitizes tumor cells to ionizing radiation. FEBS Lett. 2006, 580, 4353–4356. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Arvey, A.; Josefowicz, S.Z.; Peng, X.; Reynolds, A.; Sandstrom, R.; Neph, S.; Sabo, P.; Kim, J.M.; Liao, W.; et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 2012, 151, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Profumo, A.; Querzola, F.; Vidali, G. Core histone acetylation during lymphocyte activation. FEBS Lett. 1989, 250, 297–300. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, B.; Zhou, Z.; Hancock, W.W.; Zhang, H.; Greene, M.I. Histone acetyltransferase mediated regulation of FOXP3 acetylation and Treg function. Curr. Opin. Immunol. 2010, 22, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Van Loosdregt, J.; Vercoulen, Y.; Guichelaar, T.; Gent, Y.Y.; Beekman, J.M.; van Beekum, O.; Brenkman, A.B.; Hijnen, D.J.; Mutis, T.; Kalkhoven, E.; et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood 2010, 115, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Predina, J.; Han, R.; Beier, U.H.; Wang, L.C.; Kapoor, V.; Bhatti, T.R.; Akimova, T.; Singhal, S.; et al. Inhibition of p300 impairs Foxp3(+) T regulatory cell function and promotes antitumor immunity. Nat. Med. 2013, 19, 1173–1177. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.S.; Jang, S.W.; Kim, M.K.; Kim, L.K.; Kim, B.S.; Kim, H.S.; Kim, K.; Lee, W.; Flavell, R.A.; Lee, G.R. YY1 inhibits differentiation and function of regulatory T cells by blocking Foxp3 expression and activity. Nat. Commun. 2016, 7, 10789. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Borde, M.; Heissmeyer, V.; Feuerer, M.; Lapan, A.D.; Stroud, J.C.; Bates, D.L.; Guo, L.; Han, A.; Ziegler, S.F.; et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 2006, 126, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Bucci, J.G.; English, R.V.; Jordan, H.L.; Childers, T.A.; Tompkins, M.B.; Tompkins, W.A. Mucosally transmitted feline immunodeficiency virus induces a CD8+ antiviral response that correlates with reduction of cell-associated virus. J. Infect. Dis. 1998, 177, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Kishore, A.H.; Vedamurthy, B.M.; Mantelingu, K.; Agrawal, S.; Reddy, B.A.; Roy, S.; Rangappa, K.S.; Kundu, T.K. Specific small-molecule activator of Aurora kinase A induces autophosphorylation in a cell-free system. J. Med. Chem. 2008, 51, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Kim, E.J.; Cheng, Y.; Shin, M.H.; Oh, J.H.; Lee, D.H.; Chung, J.H. Inhibition of DNA Methylation in the COL1A2 Promoter by Anacardic Acid Prevents UV-Induced Decrease of Type I Procollagen Expression. J. Investig. Dermatol. 2017, 137, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Chen, B.; He, L.; Tang, Y.; Jiang, Z.; Yin, G.; Wang, J.; Jiang, X. Anacardic acid (6-pentadecylsalicylic acid) induces apoptosis of prostate cancer cells through inhibition of androgen receptor and activation of p53 signaling. Chin. J. Cancer Res. 2012, 24, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; He, L.; Zhang, L.; Chen, J.; Yi, Z.; Zhang, J.; Liu, M.; Pang, X. Anacardic acid (6-pentadecylsalicylic acid) inhibits tumor angiogenesis by targeting Src/FAK/Rho GTPases signaling pathway. J. Pharmacol. Exp. Ther. 2011, 339, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Xiu, Y.L.; Zhao, Y.; Gou, W.F.; Chen, S.; Takano, Y.; Zheng, H.C. Anacardic acid enhances the proliferation of human ovarian cancer cells. PLoS ONE 2014, 9, e99361. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Primer Target | Forward | Reverse |
|---|---|---|
| Foxp3 | 5’-GCCTGCCACCTGGAATCAAC-3’ | 5’-GTGTGCTGGGGCTTGGGA-3’ |
| IL-2 | 5’-ACAGTGCACCTGCTTCAAGCTCT-3’ | 5’-CCTGGAGAGTTTGGGGTTCTCAGG-3’ |
| GAPDH | 5’-GGAGAAGGCTGGGGCTCAC-3’ | 5’GGTGCAGGAGGCATTGCTGA-3’ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nag, M.; Wang, Y.; De Paris, K.; E. Fogle, J. Histone Modulation Blocks Treg-Induced Foxp3 Binding to the IL-2 Promoter of Virus-Specific CD8+ T Cells from Feline Immunodeficiency Virus-Infected Cats. Viruses 2018, 10, 287. https://doi.org/10.3390/v10060287
Nag M, Wang Y, De Paris K, E. Fogle J. Histone Modulation Blocks Treg-Induced Foxp3 Binding to the IL-2 Promoter of Virus-Specific CD8+ T Cells from Feline Immunodeficiency Virus-Infected Cats. Viruses. 2018; 10(6):287. https://doi.org/10.3390/v10060287
Chicago/Turabian StyleNag, Mukta, Yan Wang, Kristina De Paris, and Jonathan E. Fogle. 2018. "Histone Modulation Blocks Treg-Induced Foxp3 Binding to the IL-2 Promoter of Virus-Specific CD8+ T Cells from Feline Immunodeficiency Virus-Infected Cats" Viruses 10, no. 6: 287. https://doi.org/10.3390/v10060287
APA StyleNag, M., Wang, Y., De Paris, K., & E. Fogle, J. (2018). Histone Modulation Blocks Treg-Induced Foxp3 Binding to the IL-2 Promoter of Virus-Specific CD8+ T Cells from Feline Immunodeficiency Virus-Infected Cats. Viruses, 10(6), 287. https://doi.org/10.3390/v10060287