Small RNA NGS Revealed the Presence of Cherry Virus A and Little Cherry Virus 1 on Apricots in Hungary



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material, Sample Preparation
2.2. Pipeline for Data Evaluation of NGS Results (Bioinformatics)
2.3. Validation of Predicted Virus Diagnostics by RT-PCR
2.4. Phylogenetic Analysis
2.5. Validation by Northern Blot
3. Results and Discussion
3.1. Small RNA NGS Revealed the Presence of CVA and LChV-1
3.1.1. Initial Statistics
3.1.2. Small RNA NGS-Based Virus Diagnostics
3.2. Validation of the Small RNA NGS Virus Diagnostics
3.2.1. Validation of the Presence of Cherry Virus A
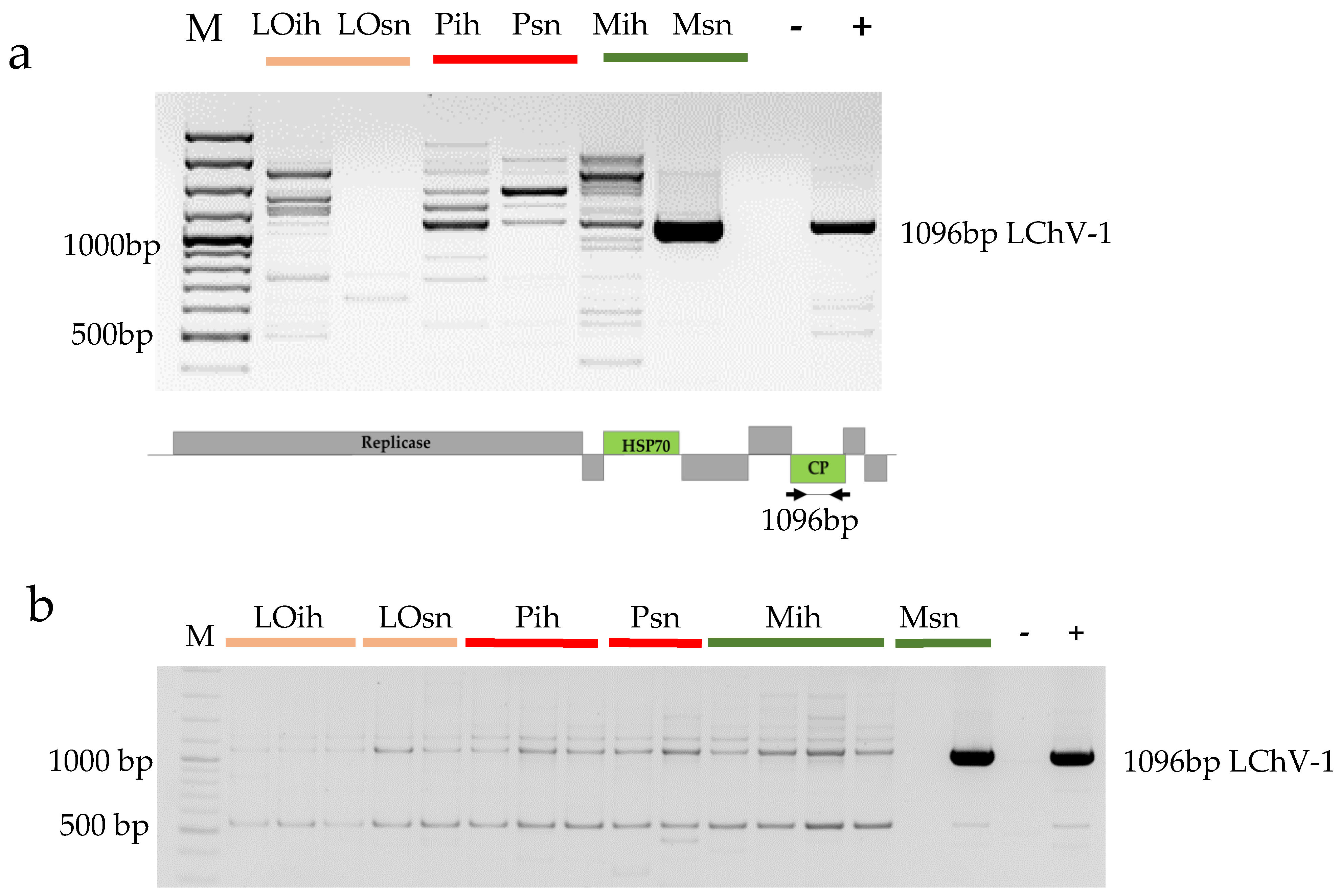
3.2.2. Validation of the Presence of Little Cherry Virus 1
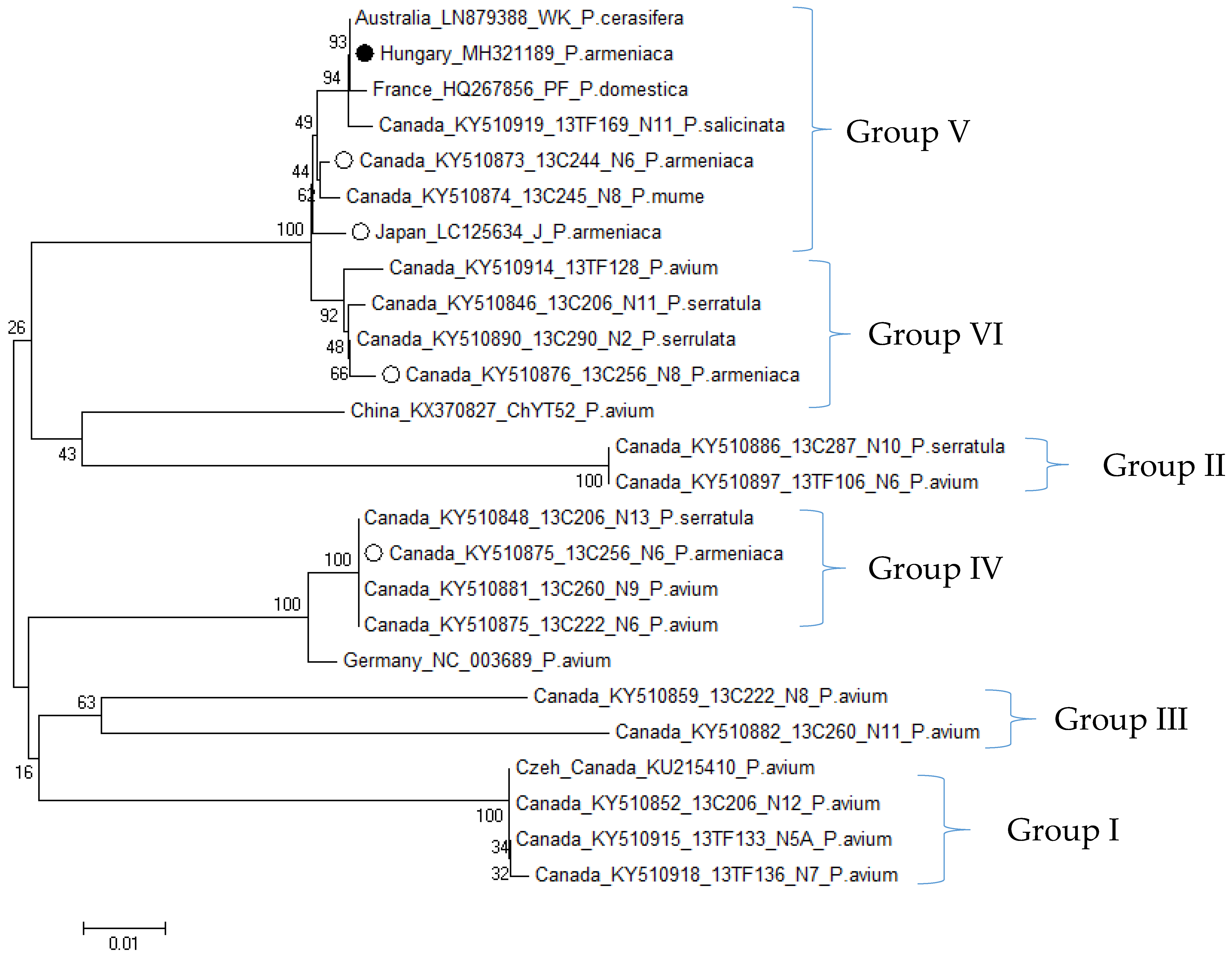
3.2.3. Phylogenetic Relationship of Hungarian CVA and LChV-1 Isolates
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Varveri, C.; Maliogka, V.I.; Kapari-Isaia, T. Chapter One—Principles for Supplying Virus-Tested Material. In Advances in Virus Research; Loebenstein, G., Katis, N.I., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 91, pp. 1–32. [Google Scholar]
- Barba, M.; Ilardi, V.; Pasquini, G. Chapter Three—Control of Pome and Stone Fruit Virus Diseases. In Advances in Virus Research; Loebenstein, G., Katis, N.I., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 91, pp. 47–83. [Google Scholar]
- Barba, M.; Czosnek, H.; Hadidi, A. Historical Perspective, Development and Applications of Next-Generation Sequencing in Plant Virology. Viruses 2014, 6, 106–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonham, N.; Kreuze, J.; Winter, S.; van der Vlugt, R.; Bergervoet, J.; Tomlinson, J.; Mumford, R. Methods in virus diagnostics: From ELISA to next generation sequencing. Virus Res. 2014, 186, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Hadidi, A.; Flores, R.; Candresse, T.; Barba, M. Next-Generation Sequencing and Genome Editing in Plant Virology. Front. Microbiol. 2016, 7, 1325. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Baizan-Edge, A.; MacFarlane, S.; Torrance, L. Viral Diagnostics in Plants Using Next Generation Sequencing: Computational Analysis in Practice. Front. Plant Sci. 2017, 8, 1770. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J. Deep sequencing for discovery and evolutionary analysis of plant viruses. Virus Res. 2017, 239, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant Virus Metagenomics: Advances in Virus Discovery. Phytopathology 2015, 105, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Elbeaino, T.; Giampetruzzi, A.; De Stradis, A.; Digiaro, M. Deep-sequencing analysis of an apricot tree with vein clearing symptoms reveals the presence of a novel betaflexivirus. Virus Res. 2014, 181, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Marais, A.; Faure, C.; Candresse, T. New Insights into Asian Prunus Viruses in the Light of NGS-Based Full Genome Sequencing. PLoS ONE 2016, 11, e0146420. [Google Scholar] [CrossRef] [PubMed]
- Marais, A.; Faure, C.; Mustafayev, E.; Barone, M.; Alioto, D.; Candresse, T. Characterization by Deep Sequencing of Prunus virus T, a Novel Tepovirus Infecting Prunus Species. Phytopathology 2014, 105, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Villamor, D.E.V.; Pillai, S.S.; Eastwell, K.C. High throughput sequencing reveals a novel fabavirus infecting sweet cherry. Arch. Virol. 2017, 162, 811–816. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Cai, L.; Zhou, L.; Yang, Z.; Hong, N.; Wang, G.; Li, S.; Xu, W. Deep sequencing reveals the first fabavirus infecting peach. Sci. Rep. 2017, 7, 11329. [Google Scholar] [CrossRef] [PubMed]
- Marais, A.; Faure, C.; Mustafayev, E.; Candresse, T. Characterization of New Isolates of Apricot vein clearing-associated virus and of a New Prunus-Infecting Virus: Evidence for Recombination as a Driving Force in Betaflexiviridae Evolution. PLoS ONE 2015, 10, e0129469. [Google Scholar] [CrossRef] [PubMed]
- Koloniuk, I.; Sarkisova, T.; Petrzik, K.; Lenz, O.; Přibylová, J.; Fránová, J.; Špak, J.; Lotos, L.; Beta, C.; Katsiani, A.; et al. Variability Studies of Two Prunus-Infecting Fabaviruses with the Aid of High-Throughput Sequencing. Viruses 2018, 10, 204. [Google Scholar] [CrossRef] [PubMed]
- Šafářová, D.; Faure, C.; Marais, A.; Suchá, J.; Paprštein, F.; Navrátil, M.; Candresse, T. First Report of Prunus virus F Infecting Sour Cherry in the Czech Republic. Plant Dis. 2017, 101, 1828. [Google Scholar] [CrossRef]
- Šafářová, D.; Faure, C.; Candresse, T.; Navrátil, M.; Nečas, T.; Marais, A. First Report of Little cherry virus 1 Infecting Apricot in the Czech Republic. Plant Dis. 2016, 101, 845. [Google Scholar] [CrossRef]
- James, D.; Phelan, J.; Jesperson, G. First Report of Prunus virus F infecting sweet cherry (Prunus avium cv. ‘StaccatoTM’) in Canada. Plant Dis. 2018. [Google Scholar] [CrossRef]
- Donaire, L.; Wang, Y.; Gonzalez-Ibeas, D.; Mayer, K.F.; Aranda, M.A.; Llave, C. Deep-sequencing of plant viral small RNAs reveals effective and widespread targeting of viral genomes. Virology 2009, 392, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parent, J.-S.; Martinez de Alba, A.E.; Vaucheret, H. The origin and effect of small RNA signaling in plants. Front. Plant Sci. 2012, 3, 179. [Google Scholar] [CrossRef] [PubMed]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecman, A.; Kutnjak, D.; Gutierrez-Aguirre, I.; Adams, I.; Fox, A.; Boonham, N.; Ravnikar, M. Next Generation Sequencing for Detection and Discovery of Plant Viruses and Viroids: Comparison of Two Approaches. Front. Microbiol. 2017, 8, 1998. [Google Scholar] [CrossRef] [PubMed]
- Santala, J.; Valkonen, J.P.T. Sensitivity of Small RNA-Based Detection of Plant Viruses. Front. Microbiol. 2018, 9, 939. [Google Scholar] [CrossRef] [PubMed]
- Navarro, B.; Pantaleo, V.; Gisel, A.; Moxon, S.; Dalmay, T.; Bisztray, G.; Di Serio, F.; Burgyan, J. Deep sequencing of viroid-derived small RNAs from grapevine provides new insights on the role of RNA silencing in plant-viroid interaction. PLoS ONE 2009, 4, e7686. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, V.; Saldarelli, P.; Miozzi, L.; Giampetruzzi, A.; Gisel, A.; Moxon, S.; Dalmay, T.; Bisztray, G.; Burgyan, J. Deep sequencing analysis of viral short RNAs from an infected Pinot Noir grapevine. Virology 2010, 408, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Giampetruzzi, A.; Roumi, V.; Roberto, R.; Malossini, U.; Yoshikawa, N.; La Notte, P.; Terlizzi, F.; Credi, R.; Saldarelli, P. A new grapevine virus discovered by deep sequencing of virus- and viroid-derived small RNAs in Cv Pinot gris. Virus Res. 2012, 163, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wang, Y.; Cao, M.; Pantaleo, V.; Burgyan, J.; Li, W.X.; Ding, S.W. Homology-independent discovery of replicating pathogenic circular RNAs by deep sequencing and a new computational algorithm. Proc. Natl. Acad. Sci. USA 2012, 109, 3938–3943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czotter, N.; Molnar, J.; Szabó, E.; Demian, E.; Kontra, L.; Baksa, I.; Szittya, G.; Kocsis, L.; Deak, T.; Bisztray, G.; et al. NGS of Virus-Derived Small RNAs as a Diagnostic Method Used to Determine Viromes of Hungarian Vineyards. Front. Microbiol. 2018, 9, 122. [Google Scholar] [CrossRef]
- Jelkmann, W. Cherry virus A: CDNA cloning of dsRNA, nucleotide sequence analysis and serology reveal a new plant capillovirus in sweet cherry. J. Gen. Virol. 1995, 76 Pt 8, 2015–2024. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Xu, Y.; Candresse, T.; He, Z.; Li, S.; Ma, Y.; Lu, M. Further insight into genetic variation and haplotype diversity of Cherry virus A from China. PLoS ONE 2017, 12, e0186273. [Google Scholar] [CrossRef] [PubMed]
- Marais, A.; Svanella, D.L.; Barone, M.; Gentit, P.; Faure, C.; Charlot, G.; Ragozzino, A.; Candresse, T. Development of a polyvalent RT-PCR detection assay covering the genetic diversity of Cherry capillovirus A. Plant Pathol. 2012, 61, 195–204. [Google Scholar] [CrossRef]
- Kesanakurti, P.; Belton, M.; Saeed, H.; Rast, H.; Boyes, I.; Rott, M. Comparative analysis of cherry virus A genome sequences assembled from deep sequencing data. Arch. Virol. 2017, 162, 2821–2828. [Google Scholar] [CrossRef] [PubMed]
- Marais, A.; Faure, C.; Svanella-Dumas, L.; Candresse, T. First Report of Cherry virus A in Prunus mume in China. Plant Dis. 2008, 92, 1589. [Google Scholar] [CrossRef]
- Keim-Konrad, R.; Jelkmann, W. Genome analysis of the 3′-terminal part of the little cherry disease associated dsRNA reveals a monopartite clostero-like virus. Arch. Virol. 1996, 141, 1437–1451. [Google Scholar] [CrossRef] [PubMed]
- Matic, S.; Minafra, A.; Sánchez-Navarro, J.A.; Pallás, V.; Myrta, A.; Martelli, G.P. ‘Kwanzan Stunting’ syndrome: Detection and molecular characterization of an Italian isolate of Little cherry virus 1. Virus Res. 2009, 143, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Candresse, T.; Marais, A.; Faure, C.; Gentit, P. Association of Little cherry virus 1 (LChV1) with the Shirofugen Stunt Disease and Characterization of the Genome of a Divergent LChV1 Isolate. Phytopathology 2013, 103, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Glasa, M. First report of little cherry virus-1 in Slovakia. J. Plant Pathol. 2015, 97, 542. [Google Scholar]
- Sabanadzovic, S.; Aboughanem, N.; Rowhani, A.; Grant, J.A.; Uyemoto, J. Detection of Cherry virus A, Cherry necrotic rusty mottle virus and Little cherry virus 1 in California orchards. J. Plant Pathol. 2005, 87, 173. [Google Scholar]
- Komorowska, B.; Cieślińska, M. First Report of Cherry virus A and Little cherry virus-1 in Poland. Plant Dis. 2004, 88, 909. [Google Scholar] [CrossRef]
- Bajet, N.B.; Unruh, T.R.; Druffel, K.L.; Eastwell, K.C. Occurrence of Two Little Cherry Viruses in Sweet Cherry in Washington State. Plant Dis. 2008, 92, 234–238. [Google Scholar] [CrossRef]
- Katsiani, A.T.; Maliogka, V.I.; Amoutzias, G.D.; Efthimiou, K.E.; Katis, N.I. Insights into the genetic diversity and evolution of Little cherry virus 1. Plant Pathol. 2015, 64, 817–824. [Google Scholar] [CrossRef]
- Czotter, N.; Molnár, J.; Pesti, R.; Demián, E.; Baráth, D.; Varga, T.; Várallyay, É. Use of siRNAs for Diagnosis of Viruses Associated to Woody Plants in Nurseries and Stock Collections. In Viral Metagenomics: Methods and Protocols; Pantaleo, V., Chiumenti, M., Eds.; Springer: New York, NY, USA, 2018; pp. 115–130. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schaffer, A.A. Database indexing for production MegaBLAST searches. Bioinformatics 2008, 24, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, T.; Candresse, T.; Ravelonandro, M.; Dunez, J. A polymerase chain reaction assay adapted to plum pox potyvirus detection. J. Virol. Methods 1991, 33, 355–365. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilks, J.M.; Welsh, M.F. Host range studies of the little cherry disease virus. Can. J. Plant Sci. 1961, 41, 544–548. [Google Scholar] [CrossRef]
- Jelkmann, W.; Fechtner, B.; Agranovsky, A.A. Complete genome structure and phylogenetic analysis of little cherry virus, a mealybug-transmissible closterovirus. J. Gen. Virol. 1997, 78, 2067–2071. [Google Scholar] [CrossRef] [PubMed]
- Rott, M.; Xiang, Y.; Boyes, I.; Belton, M.; Saeed, H.; Kesanakurti, P.; Hayes, S.; Lawrence, T.; Birch, C.; Bhagwat, B.; et al. Application of Next Generation Sequencing for Diagnostic Testing of Tree Fruit Viruses and Viroids. Plant Dis. 2017, 101, 1489–1499. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Islas, C.; Rowhani, A. Comparison of Next-Generation Sequencing Versus Biological Indexing for the Optimal Detection of Viral Pathogens in Grapevine. Phytopathology 2015, 105, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Massart, S.; Candresse, T.; Gil, J.; Lacomme, C.; Predajna, L.; Ravnikar, M.; Reynard, J.-S.; Rumbou, A.; Saldarelli, P.; Škorić, D.; et al. A Framework for the Evaluation of Biosecurity, Commercial, Regulatory, and Scientific Impacts of Plant Viruses and Viroids Identified by NGS Technologies. Front. Microbiol. 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Massart, S.; Olmos, A.; Jijakli, H.; Candresse, T. Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 2014, 188, 90–96. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variety | Type of Analysis | Origin of Samples | ||
|---|---|---|---|---|
| In Vitro | Isolator House | Stock Nursery | ||
| Ligeti óriás | NGS virus hit | - | 0 | PPV |
| PCR * | 0 | 0 | 1/2 PPV | |
| Pannónia | NGS virus hit | - | CVA | CVA |
| PCR * | 2/4 CVA | 3/3 CVA | 2/2 CVA | |
| Magyar kajszi | NGS virus hit | - | 0 | PPV, LChV-1 |
| PCR * | 0 | 0 | 1/2 PPV, 1/2 LChV-1 | |
| Virus | Variety | Genebank Identifier | Genome Used as a Reference | Position on the Reference Genome | Function of the Amplified Part of the Genome | Identity on Nucleotide Level (%) | Identity on Amino Acid Level (%) |
|---|---|---|---|---|---|---|---|
| CVA | Pannónia kajszi/Pannonian apricot | MH321189 | NC_003689.1 | 5401–6791 | replicase (partial) | 1275/1392(92%) | 392/463(85%) |
| 5400–6791 | movement protein | 421/463(91%) | |||||
| LChV-1 | Magyar kajszi/Hungarian apricot | MH321190 | NC_001836.1 | 12,165–13,261 | coat protein | 1011/1097(92%) | 338/365(93%) |
| LChV-1 | Magyar kajszi/Hungarian apricot | MH321191 | NC_001836.1 | 9493–10,288 | HSP70h | 753/796(95%) | 251/265(95%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baráth, D.; Jaksa-Czotter, N.; Molnár, J.; Varga, T.; Balássy, J.; Szabó, L.K.; Kirilla, Z.; Tusnády, G.E.; Preininger, É.; Várallyay, É. Small RNA NGS Revealed the Presence of Cherry Virus A and Little Cherry Virus 1 on Apricots in Hungary. Viruses 2018, 10, 318. https://doi.org/10.3390/v10060318
Baráth D, Jaksa-Czotter N, Molnár J, Varga T, Balássy J, Szabó LK, Kirilla Z, Tusnády GE, Preininger É, Várallyay É. Small RNA NGS Revealed the Presence of Cherry Virus A and Little Cherry Virus 1 on Apricots in Hungary. Viruses. 2018; 10(6):318. https://doi.org/10.3390/v10060318
Chicago/Turabian StyleBaráth, Dániel, Nikoletta Jaksa-Czotter, János Molnár, Tünde Varga, Júlia Balássy, Luca Krisztina Szabó, Zoltán Kirilla, Gábor E. Tusnády, Éva Preininger, and Éva Várallyay. 2018. "Small RNA NGS Revealed the Presence of Cherry Virus A and Little Cherry Virus 1 on Apricots in Hungary" Viruses 10, no. 6: 318. https://doi.org/10.3390/v10060318
APA StyleBaráth, D., Jaksa-Czotter, N., Molnár, J., Varga, T., Balássy, J., Szabó, L. K., Kirilla, Z., Tusnády, G. E., Preininger, É., & Várallyay, É. (2018). Small RNA NGS Revealed the Presence of Cherry Virus A and Little Cherry Virus 1 on Apricots in Hungary. Viruses, 10(6), 318. https://doi.org/10.3390/v10060318