Molecular Aspects of Varicella-Zoster Virus Latency


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular Biology of VZV
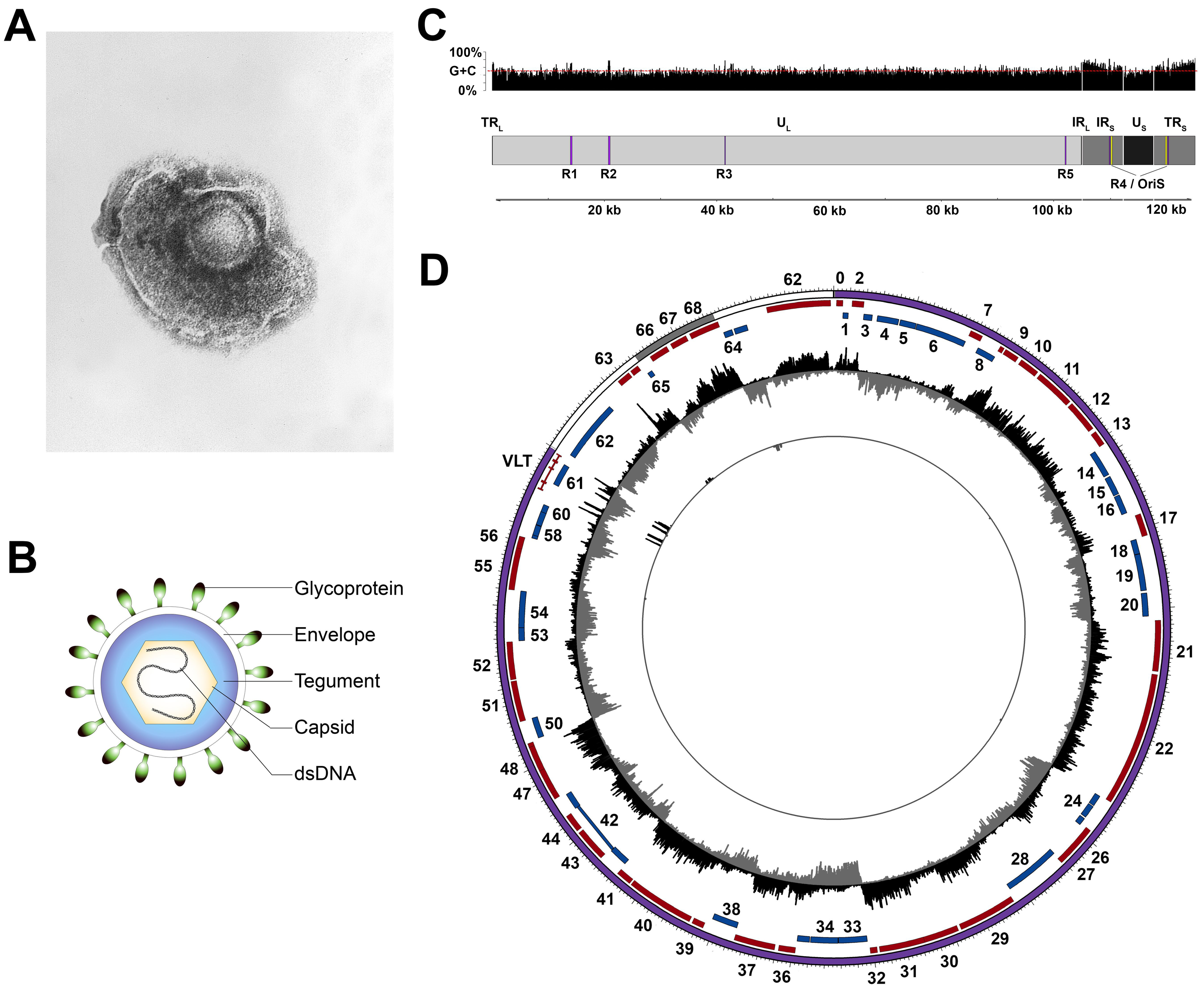
2.1. Structure and Genomic Organization of VZV
2.2. Coding Potential of the VZV Genome
2.3. VZV Gene Expression during Productive Infection
2.4. Stability of the VZV Genome
3. Location of Latent VZV
3.1. Sites of VZV Latency
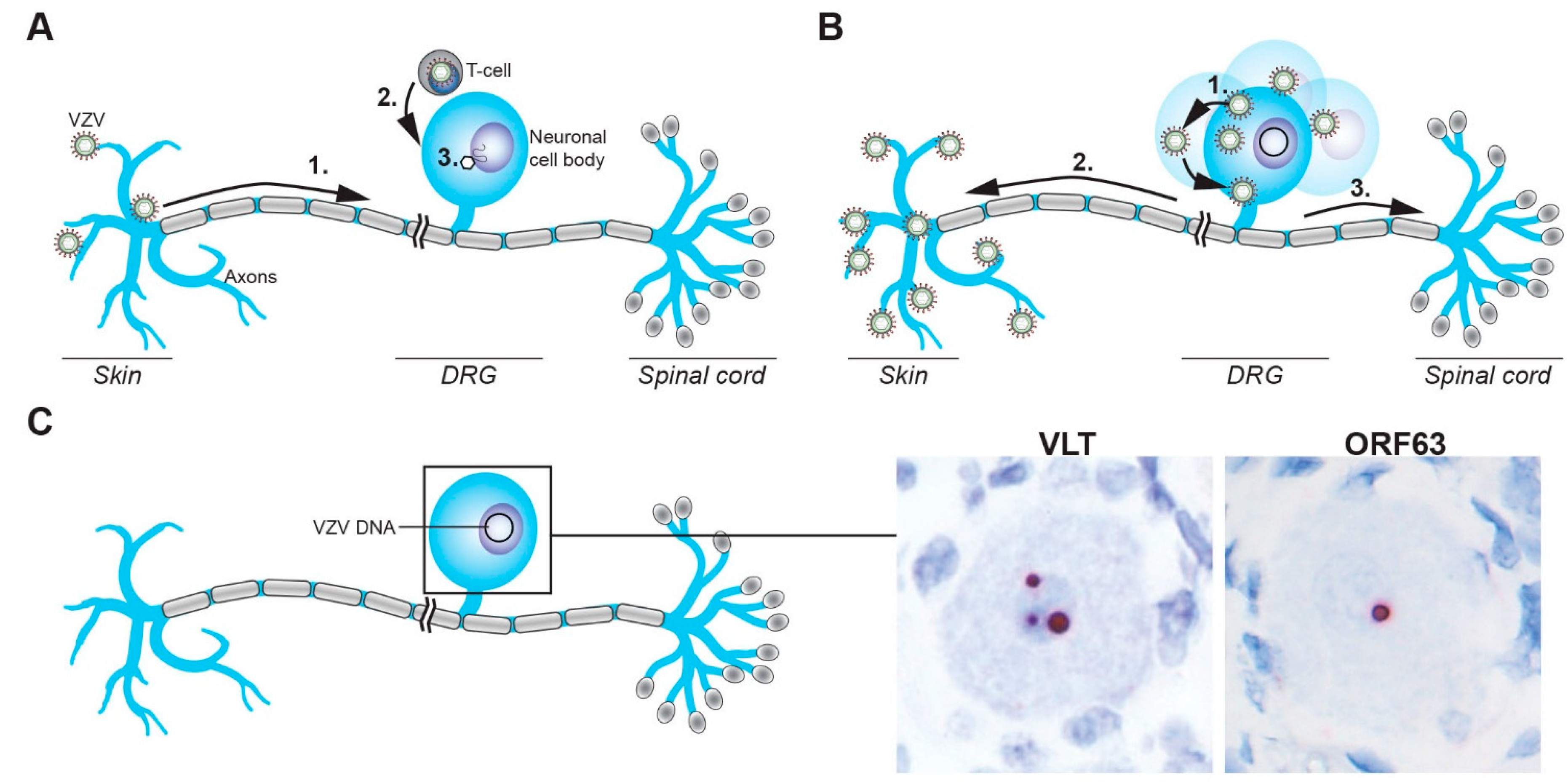
3.2. Entry of VZV into the Peripheral Nervous System
4. Transcriptional Repression of Latent VZV Genomes
4.1. VZV Transcription in Human Ganglia
4.2. Comparison of VZV Latency In Vivo and In Vitro
4.3. Epigenetic Silencing of the Latent VZV Genome
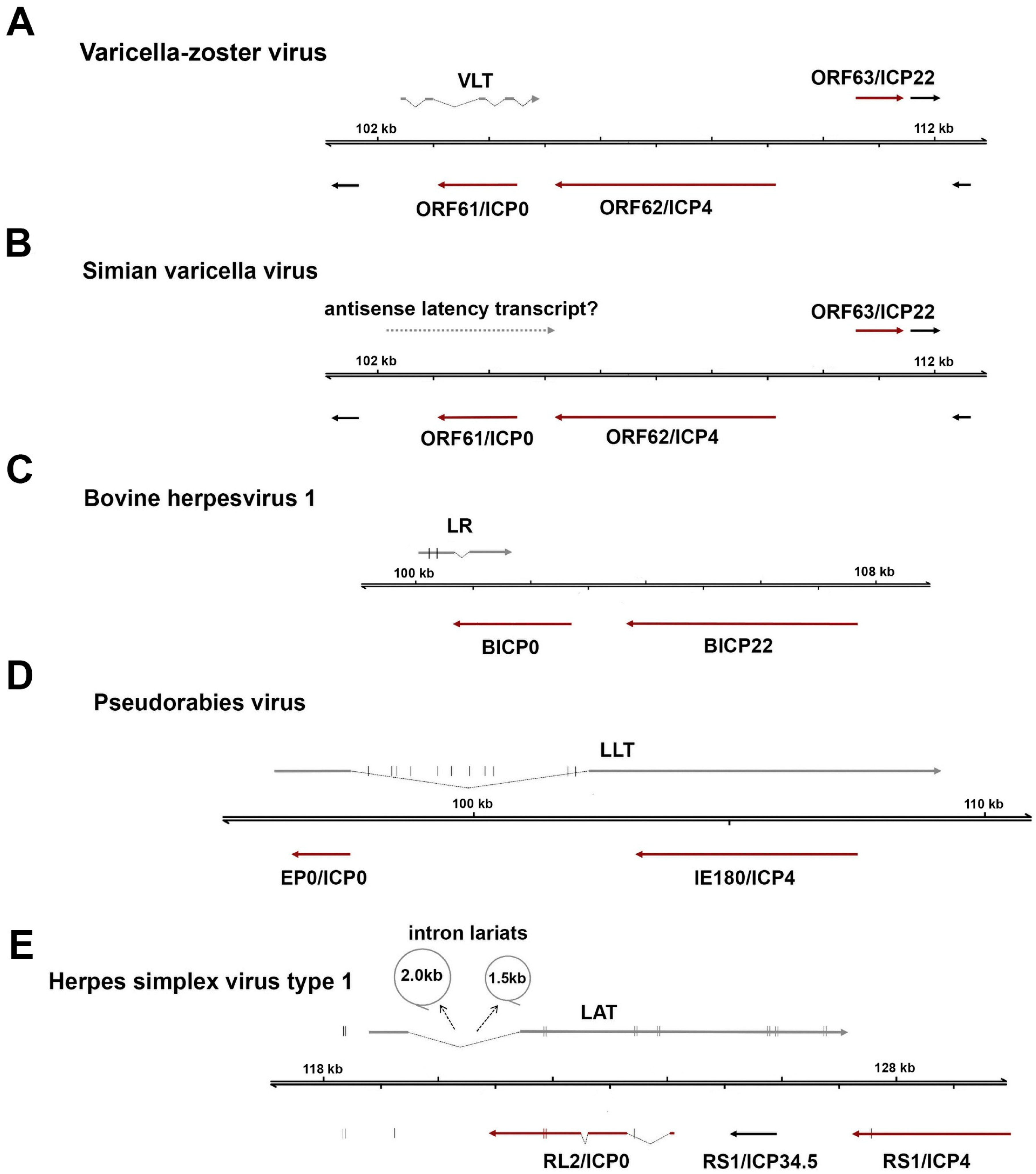
5. The Varicella Zoster Virus Latency-Associated Transcripts: VLT and ORF63
5.1. Comparison of VLT and LATs of Related Alphaherpesviruses
5.2. Function(s) of VLT
5.3. Function(s) of ORF63
6. VZV Reactivation: Restarting Lytic Gene Expression
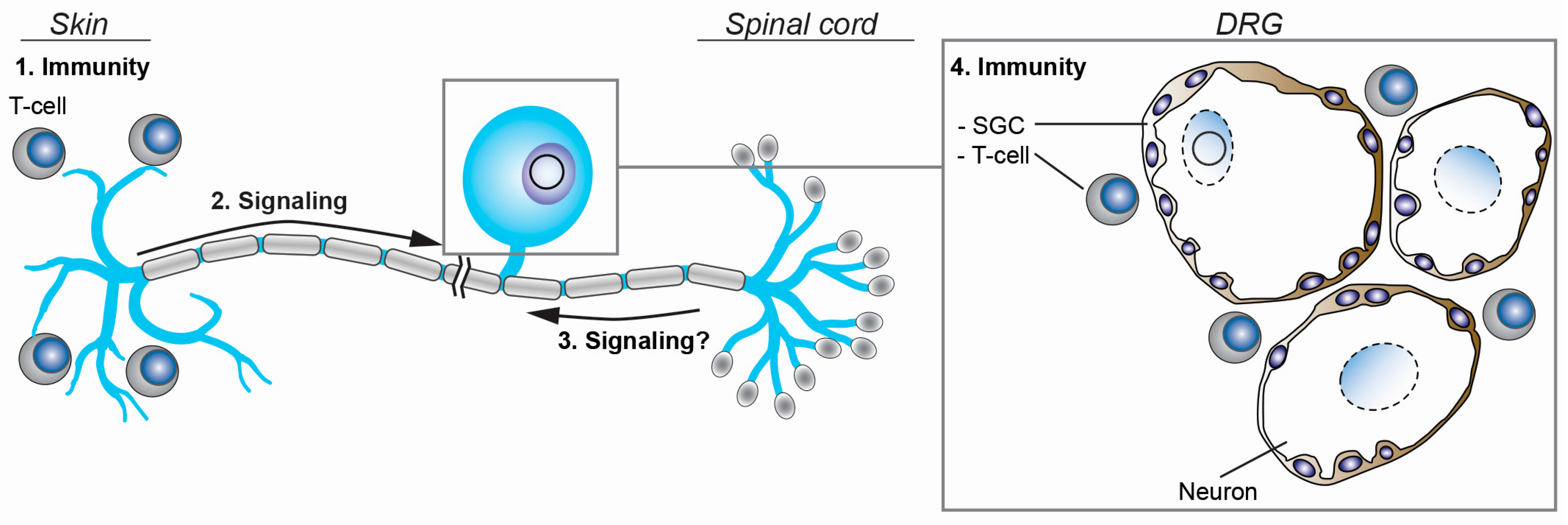
7. Intrinsic, Innate, and Adaptive Immunity to VZV Infection in Ganglia
7.1. Satellite Glial Cells and Innate Immunity
7.2. T-Cell Immunity
8. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Virgin, H.W.; Wherry, E.J.; Ahmed, R. Redefining Chronic Viral Infection. Cell 2009, 138, 30–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershon, A.A.; Breuer, J.; Cohen, J.I.; Cohrs, R.J.; Gershon, M.D.; Gilden, D.; Grose, C.; Hambleton, S.; Kennedy, P.G.E.; Oxman, M.N.; et al. Varicella zoster virus infection. Nat. Rev. Dis. Prim. 2015, 1, 15016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, T.H.; Witton, H.M.; Bell, E.J. The etiologic agents of varicella and herpes zoster. J. Exp. Med. 1958, 108, 843–868. [Google Scholar] [CrossRef] [PubMed]
- Hope-Simpson, R.E. The Nature of Herpes Zoster: A Long-term Study and a New Hypothesis. Proc. R. Soc. Med. 1965, 58, 9–20. [Google Scholar] [PubMed]
- Gilden, D.H.; Vafai, A.; Shtram, Y.; Becker, Y.; Devlin, M.; Wellish, M. Varicella-zoster virus DNA in human sensory ganglia. Nature 1983, 306, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Padilla, J.A.; Nii, S.; Grose, C. Imaging of the varicella zoster virion in the viral highways: Comparison with herpes simplex viruses 1 and 2, cytomegalovirus, pseudorabies virus, and human herpes viruses 6 and 7. J. Med. Virol. 2003, 70 (Suppl. 1), S103–S110. [Google Scholar] [CrossRef] [PubMed]
- Arvin, A.M.; Gilden, D. Varicella-Zoster Virus. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2015–2057. [Google Scholar]
- Storlie, J.; Maresova, L.; Jackson, W.; Grose, C. Comparative Analyses of the 9 Glycoprotein Genes Found in Wild-Type and Vaccine Strains of Varicella-Zoster Virus. J. Infect. Dis. 2008, 197, S49–S53. [Google Scholar] [CrossRef] [PubMed]
- Penkert, R.R.; Kalejta, R.F. Tegument protein control of latent herpesvirus establishment and animation. Herpesviridae 2011, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, H.; Haines, H.G.; Biswal, N.; Benyesh-Melnick, M. The Characterization of Varicella-zoster Virus DNA. J. Gen. Virol. 1972, 14, 111–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakes, J.E.; Iltis, J.P.; Hyman, R.W.; Rapp, F. Analysis by restriction enzyme cleavage of human varicella-zoster virus DNAs. Virology 1977, 82, 353–361. [Google Scholar] [CrossRef]
- Richards, J.C.; Hyman, R.W.; Rapp, F. Analysis of the DNAs from seven varicella-zoster virus isolates. J. Virol. 1979, 32, 812–821. [Google Scholar] [PubMed]
- Zweerink, H.J.; Morton, D.H.; Stanton, L.W.; Neff, B.J. Restriction endonuclease analysis of the DNA from varicella-zoster virus: Stability of the DNA after passage in vitro. J. Gen. Virol. 1981, 55, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Straus, S.E.; Aulakh, H.S.; Ruyechan, W.T.; Hay, J.; Casey, T.A.; Vande Woude, G.F.; Owens, J.; Smith, H.A. Structure of varicella-zoster virus DNA. J. Virol. 1981, 40, 516–525. [Google Scholar] [PubMed]
- Straus, S.E.; Owens, J.; Ruyechan, W.T.; Takiff, H.E.; Casey, T.A.; Vande Woude, G.F.; Hay, J. Molecular cloning and physical mapping of varicella-zoster virus DNA. Proc. Natl. Acad. Sci. USA 1982, 79, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.H.; Dohner, D.E.; Wellinghoff, W.J.; Gelb, L.D. Restriction endonuclease analysis of varicella-zoster vaccine virus and wild-type dnas. J. Med. Virol. 1982, 9, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.M.; Geelen, J.L.; Weststrate, M.W.; Wertheim, P.; van der Noordaa, J. XbaI, PstI, and BglII restriction enzyme maps of the two orientations of the varicella-zoster virus genome. J. Virol. 1981, 39, 390–400. [Google Scholar] [PubMed]
- Dumas, A.M.; Geelen, J.L.M.C.; Maris, W.; van der Noordaa, J. Infectivity and Molecular Weight of Varicella-Zoster Virus DNA. J. Gen. Virol. 1980, 47, 233–235. [Google Scholar] [CrossRef] [Green Version]
- Davison, A.J.; Scott, J.E. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 1986, 67, 1759–1816. [Google Scholar] [CrossRef] [PubMed]
- Kinchington, P.R.; Reinhold, W.C.; Casey, T.A.; Straus, S.E.; Hay, J.; Ruyechan, W.T. Inversion and circularization of the varicella-zoster virus genome. J. Virol. 1985, 56, 194–200. [Google Scholar] [PubMed]
- Kaufer, B.B.; Smejkal, B.; Osterrieder, N. The varicella-zoster virus ORFS/L (ORF0) gene is required for efficient viral replication and contains an element involved in DNA cleavage. J. Virol. 2010, 84, 11661–11669. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Hondo, R.; Taguchi, F.; Yogo, Y. Variation of R1 repeated sequence present in open reading frame 11 of varicella-zoster virus strains. J. Virol. 1988, 62, 1097–1100. [Google Scholar] [PubMed]
- Hondo, R.; Yogo, Y. Strain variation of R5 direct repeats in the right-hand portion of the long unique segment of varicella-zoster virus DNA. J. Virol. 1988, 62, 2916–2921. [Google Scholar] [PubMed]
- Sauerbrei, A.; Zell, R.; Wutzler, P. Analysis of repeat units in the R2 region among different Oka varicella-zoster virus vaccine strains and wild-type strains in Germany. Intervirology 2006, 50, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Gray, E.R.; Kundu, S.; Cooray, S.; Poulsen, A.; Aaby, P.; Breuer, J. Evolution of cocirculating varicella-zoster virus genotypes during a chickenpox outbreak in Guinea-Bissau. J. Virol. 2014, 88, 13936–13946. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Tamura, T.; Miyasaka, K.; Shimizu, A.; Ohashi, N.; Itoh, M. Analysis of numbers of repeated units in R2 region among varicella-zoster virus strains. J. Dermatol. Sci. 2003, 31, 129–133. [Google Scholar] [CrossRef]
- Depledge, D.P.; Ouwendijk, W.J.D.; Sadaoka, T.; Braspenning, S.E.; Mori, Y.; Cohrs, R.J.; Verjans, G.M.G.M.; Breuer, J. A spliced latency-associated VZV transcript maps antisense to the viral transactivator gene 61. Nat. Commun. 2018, 9, 1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemble, G.W.; Annunziato, P.; Lungu, O.; Winter, R.E.; Cha, T.A.; Silverstein, S.J.; Spaete, R.R. Open reading frame S/L of varicella-zoster virus encodes a cytoplasmic protein expressed in infected cells. J. Virol. 2000, 74, 11311–11321. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Williams, M.; Cohen, J.I. Disruption of the varicella-zoster virus dUTPase and the adjacent ORF9A gene results in impaired growth and reduced syncytia formation in vitro. Virology 1997, 234, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Preston, V.G.; Kennard, J.; Rixon, F.J.; Logan, A.J.; Mansfield, R.W.; McDougall, I.M. Efficient herpes simplex virus type 1 (HSV-1) capsid formation directed by the varicella-zoster virus scaffolding protein requires the carboxy-terminal sequences from the HSV-1 homologue. J. Gen. Virol. 1997, 78, 1633–1646. [Google Scholar] [CrossRef] [PubMed]
- Sadaoka, T.; Yanagi, T.; Yamanishi, K.; Mori, Y. Characterization of the varicella-zoster virus ORF50 gene, which encodes glycoprotein M. J. Virol. 2010, 84, 3488–3502. [Google Scholar] [CrossRef] [PubMed]
- Koshizuka, T.; Ota, M.; Yamanishi, K.; Mori, Y. Characterization of varicella-zoster virus-encoded ORF0 gene-Comparison of parental and vaccine strains. Virology 2010, 405, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.A.; Tyler, S.D.; Carpenter, J.E.; Jackson, W.; Mori, Y.; Arvin, A.M.; Grose, C. The attenuated genotype of varicella-zoster virus includes an ORF0 transitional stop codon mutation. J. Virol. 2012, 86, 10695–10703. [Google Scholar] [CrossRef] [PubMed]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RN A sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Nagel, M.A.; Cohrs, R.J.; Gilden, D.H.; Cullen, B.R. Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. J. Virol. 2009, 83, 10677–10683. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Golani, L.; Ojha, N.K.; Borodiansky-Shteinberg, T.; Kinchington, P.R.; Goldstein, R.S. Varicella-Zoster Virus Expresses Multiple Small Noncoding RNAs. J. Virol. 2017, 91, e01710-17. [Google Scholar] [CrossRef] [PubMed]
- Reichelt, M.; Brady, J.; Arvin, A.M. The replication cycle of varicella-zoster virus: Analysis of the kinetics of viral protein expression, genome synthesis, and virion assembly at the single-cell level. J. Virol. 2009, 83, 3904–3918. [Google Scholar] [CrossRef] [PubMed]
- Roviš, T.L.; Bailer, S.M.; Pothineni, V.R.; Ouwendijk, W.J.D.; Šimić, H.; Babić, M.; Miklić, K.; Malić, S.; Verweij, M.C.; Baiker, A.; et al. Comprehensive analysis of varicella-zoster virus proteins using a new monoclonal antibody collection. J. Virol. 2013, 87, 6943–6954. [Google Scholar] [CrossRef] [PubMed]
- Kinchington, P.R.; Hougland, J.K.; Arvin, A.M.; Ruyechan, W.T.; Hay, J. The varicella-zoster virus immediate-early protein IE62 is a major component of virus particles. J. Virol. 1992, 66, 359–366. [Google Scholar] [PubMed]
- Kinchington, P.R.; Bookey, D.; Turse, S.E. The transcriptional regulatory proteins encoded by varicella-zoster virus open reading frames (ORFs) 4 and 63, but not ORF 61, are associated with purified virus particles. J. Virol. 1995, 69, 4274–4282. [Google Scholar] [PubMed]
- Perera, L.P.; Mosca, J.D.; Sadeghi-Zadeh, M.; Ruyechan, W.T.; Hay, J. The Varicella-Zoster virus immediate early protein, IE62, can positively regulate its cognate promoter. Virology 1992, 191, 346–354. [Google Scholar] [CrossRef]
- Perera, L.P.; Mosca, J.D.; Ruyechan, W.T.; Hayward, G.S.; Straus, S.E.; Hay, J. A major transactivator of varicella-zoster virus, the immediate-early protein IE62, contains a potent N-terminal activation domain. J. Virol. 1993, 67, 4474–4483. [Google Scholar] [PubMed]
- Tyler, J.T.; Allen, K.E.; Everett, R.D. Mutation of a single lysine residue severely impairs the DNA recognition and regulatory functions of the VZV gene 62 transactivator protein. Nucleic Acids Res. 1994, 22, 270–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriuchi, M.; Moriuchi, H.; Straus, S.E.; Cohen, J.I. Varicella-Zoster Virus (VZV) Virion-Associated Transactivator Open Reading Frame 62 Protein Enhances the Infectivity of VZV DNA. Virology 1994. [Google Scholar] [CrossRef] [PubMed]
- Kinchington, P.R.; Turse, S.E. Regulated nuclear localization of the varicella-zoster virus major regulatory protein, IE62. J. Infect. Dis. 1998, 178 (Suppl. 1), S16–S21. [Google Scholar] [CrossRef] [PubMed]
- Ruyechan, W.T.; Peng, H.; Yang, M.; Hay, J. Cellular factors and IE62 activation of VZV promoters. J. Med. Virol. 2003, 70, S90–S94. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Peng, H.; Hay, J.; Ruyechan, W.T. Promoter activation by the varicella-zoster virus major transactivator IE62 and the cellular transcription factor USF. J. Virol. 2006, 80, 7339–7353. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.I.; Che, X.; Sung, P.; Sommer, M.H.; Hay, J.; Arvin, A.M. Mutational analysis of varicella-zoster virus (VZV) immediate early protein (IE62) subdomains and their importance in viral replication. Virology 2016, 492, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sommer, M.; Rajamani, J.; Arvin, A.M. Regulation of the ORF61 Promoter and ORF61 Functions in Varicella-Zoster Virus Replication and Pathogenesis. J. Virol. 2009, 83, 7560–7572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, E.J.; Kuck, K.M.; Kinchington, P.R. Anatomy of the varicella-zoster virus open-reading frame 4 promoter. J. Infect. Dis. 1998, 178 (Suppl. 1), S27–S33. [Google Scholar] [CrossRef] [PubMed]
- Kost, R.G.; Kupinsky, H.; Straus, S.E. Varicella-Zoster Virus Gene 63: Transcript Mapping and Regulatory Activity. Virology 1995, 209, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Ruyechan, W.T. Roles of cellular transcription factors in VZV replication. Curr. Top. Microbiol. Immunol. 2010, 342, 43–65. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. The Varicella-Zoster Virus Genome. Curr. Top. Microbiol. Immunol. 2010, 342, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, N.L.; Bowlin, J.L.; Cohrs, R.J.; Gilden, D.; Jones, K.L. Comparison of varicella-zoster virus RNA sequences in human neurons and fibroblasts. J. Virol. 2014, 88, 5877–5880. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Waldman Ben-Asher, H.; Kinchington, P.R.; Goldstein, R.S. Cellular Transcriptome Analysis Reveals Differential Expression of Pro- and Antiapoptosis Genes by Varicella-Zoster Virus-Infected Neurons and Fibroblasts. J. Virol. 2014, 88, 7674–7677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadaoka, T.; Depledge, D.P.; Rajbhandari, L.; Venkatesan, A.; Breuer, J.; Cohen, J.I. In vitro system using human neurons demonstrates that varicella-zoster vaccine virus is impaired for reactivation, but not latency. Proc. Natl. Acad. Sci. USA 2016, 113, E2403–E2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.; Dry, I.R.; Frampton, D.; Singh, M.; Kanda, R.K.; Yee, M.B.; Kellam, P.; Hollinshead, M.; Kinchington, P.R.; O’Toole, E.A.; et al. RNA-seq Analysis of Host and Viral Gene Expression Highlights Interaction between Varicella Zoster Virus and Keratinocyte Differentiation. PLoS Pathog. 2014, 10, e1003896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depledge, D.P.; Palser, A.L.; Watson, S.J.; Lai, I.Y.C.; Gray, E.R.; Grant, P.; Kanda, R.K.; Leproust, E.; Kellam, P.; Breuer, J. Specific Capture and Whole-Genome Sequencing of Viruses from Clinical Samples. PLoS ONE 2011, 6, e27805. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Kundu, S.; Jensen, N.J.; Gray, E.R.; Jones, M.; Steinberg, S.; Gershon, A.; Kinchington, P.R.; Schmid, D.S.; Balloux, F.; et al. Deep sequencing of viral genomes provides insight into the evolution and pathogenesis of varicella zoster virus and its vaccine in humans. Mol. Biol. Evol. 2014, 31, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Norberg, P.; Depledge, D.P.; Kundu, S.; Atkinson, C.; Brown, J.; Haque, T.; Hussaini, Y.; MacMahon, E.; Molyneaux, P.; Papaevangelou, V.; et al. Recombination of Globally Circulating Varicella Zoster Virus. J. Virol. 2015, 89, 7133–7146. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Brown, J.; Macanovic, J.; Underhill, G.; Breuer, J. Viral Genome Sequencing Proves Nosocomial Transmission of Fatal Varicella. J. Infect. Dis. 2016, 214, 1399–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinert, L.A.; Depledge, D.P.; Kundu, S.; Gershon, A.A.; Nichols, R.A.; Balloux, F.; Welch, J.J.; Breuer, J. Rates of vaccine evolution show strong effects of latency: Implications for varicella zoster virus epidemiology. Mol. Biol. Evol. 2015, 32, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Taudien, S.; Pfaff, F.; Wutzler, P.; Platzer, M.; Sauerbrei, A. Sequencing of 21 Varicella-Zoster Virus Genomes Reveals Two Novel Genotypes and Evidence of Recombination. J. Virol. 2012, 86, 1608–1622. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.A.; Tyler, S.D.; Grose, C.; Severini, A.; Gray, M.J.; Upton, C.; Tipples, G.A. A Full-Genome Phylogenetic Analysis of Varicella-Zoster Virus Reveals a Novel Origin of Replication-Based Genotyping Scheme and Evidence of Recombination between Major Circulating Clades. J. Virol. 2006, 80, 9850–9860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyler, S.D.; Peters, G.A.; Grose, C.; Severini, A.; Gray, M.J.; Upton, C.; Tipples, G.A. Genomic cartography of varicella-zoster virus: A complete genome-based analysis of strain variability with implications for attenuation and phenotypic differences. Virology 2007, 359, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Takasu, T.; Fukuda, S.; Sato-Matsumura, K.C.; Inuyama, Y.; Hondo, R.; Nagashima, K. Detection of varicella-zoster virus DNA in human geniculate ganglia by polymerase chain reaction. J. Infect. Dis. 1992, 166, 1157–1159. [Google Scholar] [CrossRef] [PubMed]
- Gilden, D.H.; Rozenman, Y.; Murray, R.; Devlin, M.; Vafai, A. Detection of varicella-zoster virus nucleic acid in neurons of normal human thoracic ganglia. Ann. Neurol. 1987, 22, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Takasu, T.; Suzuki, S.; Fukuda, S.; Inuyama, Y.; Nagashima, K. Detection of latent varicella-zoster virus infection in human vestibular and spiral ganglia. J. Med. Virol. 1997, 51, 214–216. [Google Scholar] [CrossRef]
- Gershon, A.A.; Chen, J.; Gershon, M.D. A Model of Lytic, Latent, and Reactivating Varicella-Zoster Virus Infections in Isolated Enteric Neurons. J. Infect. Dis. 2008, 197, S61–S65. [Google Scholar] [CrossRef] [PubMed]
- Gilden, D.H.; Gesser, R.; Smith, J.; Wellish, M.; Laguardia, J.J.; Cohrs, R.J.; Mahalingam, R. Presence of VZV and HSV-1 DNA in human nodose and celiac ganglia. Virus Genes 2001, 23, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Nagel, M.A.; Rempel, A.; Huntington, J.; Kim, F.; Choe, A.; Gilden, D. Frequency and abundance of alphaherpesvirus DNA in human thoracic sympathetic ganglia. J. Virol. 2014, 88, 8189–8192. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.; Beer, T.; Cohrs, R.; Gilden, D.H. Configuration of latent varicella-zoster virus DNA. J. Virol. 1995, 69, 8151–8154. [Google Scholar] [PubMed]
- Kennedy, P.G.; Grinfeld, E.; Gow, J.W. Latent varicella-zoster virus is located predominantly in neurons in human trigeminal ganglia. Proc. Natl. Acad. Sci. USA 1998, 95, 4658–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, M.J.; Cai, G.Y.; Manchak, M.D.; Pizer, L.I. Varicella-zoster virus DNA in cells isolated from human trigeminal ganglia. J. Virol. 2003, 77, 6979–6987. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Lau, T.Y.; Morales, M.; Mont, E.K.; Straus, S.E. Laser-Capture Microdissection: Refining Estimates of the Quantity and Distribution of Latent Herpes Simplex Virus 1 and Varicella-Zoster Virus DNA in Human Trigeminal Ganglia at the Single-Cell Level. Cancer Biol. Ther. 2005, 79, 14079–14087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershon, A.A.; Chen, J.; Davis, L.; Krinsky, C.; Cowles, R.; Reichard, R.; Gershon, M. Latency of varicella zoster virus in dorsal root, cranial, and enteric Ganglia in vaccinated children. Trans. Am. Clin. Climatol. Assoc. 2012, 123, 17–35. [Google Scholar] [PubMed]
- Chen, J.J.; Gershon, A.A.; Li, Z.; Cowles, R.A.; Gershon, M.D. Varicella zoster virus (VZV) infects and establishes latency in enteric neurons. J. Neurovirol. 2011, 17, 578–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouwendijk, W.; van Veen, S.; Mehraban, T.; Mahalingam, R.; Verjans, G. Simian Varicella Virus Infects Enteric Neurons and α4β7 Integrin-Expressing Gut-Tropic T-Cells in Nonhuman Primates. Viruses 2018, 10, 156. [Google Scholar] [CrossRef] [PubMed]
- Gershon, A.A.; Chen, J.; Gershon, M.D. Use of Saliva to Identify Varicella Zoster Virus Infection of the Gut. Clin. Infect. Dis. 2015, 61, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrascosa, M.F.; Salcines-Caviedes, J.R.; Gómez Román, J.; Cano-Hoz, M.; Fernández-Ayala, M.; Casuso-Sáenz, E.; Abascal-Carrera, I.; Campo-Ruiz, A.; Martín, M.C.; Díaz-Pérez, A.; et al. Varicella-zoster virus (VZV) infection as a possible cause of Ogilvie’s syndrome in an immunocompromised host. J. Clin. Microbiol. 2014, 52, 2718–2721. [Google Scholar] [CrossRef] [PubMed]
- Pui, J.C.; Furth, E.E.; Minda, J.; Montone, K.T. Demonstration of varicella-zoster virus infection in the muscularis propria and myenteric plexi of the colon in an HIV-positive patient with herpes zoster and small bowel pseudo-obstruction (Ogilvie’s syndrome). Am. J. Gastroenterol. 2001, 96, 1627–1630. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, P.W.; Lungu, O.; Panagiotidis, C.; Zhang, J.H.; Silvers, D.N.; Gershon, A.A.; Silverstein, S.J. Varicella-zoster virus proteins in skin lesions: Implications for a novel role of ORF29p in chickenpox. J. Virol. 2000, 74, 2005–2010. [Google Scholar] [CrossRef] [PubMed]
- Hardy, I.; Gershon, A.A.; Steinberg, S.P.; LaRussa, P.; Varicella Vaccine Collaborative Study Group. The Incidence of Zoster after Immunization with Live Attenuated Varicella Vaccine. N. Engl. J. Med. 1991, 325, 1545–1550. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Grigoryan, S.; Sloutskin, A.; Yee, M.B.; Zhu, H.; Yang, I.H.; Thakor, N.V.; Sarid, R.; Kinchington, P.R.; Goldstein, R.S. Varicella-zoster virus (VZV) infection of neurons derived from human embryonic stem cells: Direct demonstration of axonal infection, transport of VZV, and productive neuronal infection. J. Virol. 2011, 85, 6220–6233. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, S.; Kinchington, P.R.; Yang, I.H.; Selariu, A.; Zhu, H.; Yee, M.; Goldstein, R.S. Retrograde axonal transport of VZV: Kinetic studies in hESC-derived neurons. J. Neurovirol. 2012, 18, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, S.; Yee, M.B.; Glick, Y.; Gerber, D.; Kepten, E.; Garini, Y.; Yang, I.H.; Kinchington, P.R.; Goldstein, R.S. Direct transfer of viral and cellular proteins from varicella-zoster virus-infected non-neuronal cells to human axons. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.I.; Peltier, A.C.; Li, J. Evaluating dermal myelinated nerve fibers in skin biopsy. Muscle Nerve 2013, 47, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Che, X.; Rajamani, J.; Zerboni, L.; Sung, P.; Ptacek, J.; Arvin, A.M. Signal transducer and activator of transcription 3 (STAT3) and survivin induction by varicella-zoster virus promote replication and skin pathogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.-C.; Padilla, J.A.; Grose, C.; Butcher, E.C.; Arvin, A.M. Tropism of varicella-zoster virus for human tonsillar CD4(+) T lymphocytes that express activation, memory, and skin homing markers. J. Virol. 2002, 76, 11425–11433. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.-C.; Zerboni, L.; Ito, H.; Graham, B.S.; Wallace, M.; Arvin, A.M. Varicella-zoster virus transfer to skin by T Cells and modulation of viral replication by epidermal cell interferon-alpha. J. Exp. Med. 2004, 200, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Mukherjee, G.; Sen, A.; Bendall, S.C.; Sung, P.; Nolan, G.P.; Arvin, A.M. Single-Cell Mass Cytometry Analysis of Human Tonsil T Cell Remodeling by Varicella Zoster Virus. Cell Rep. 2014, 8, 633–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerboni, L.; Ku, C.; Jones, C.D.; Zehnder, J.L.; Arvin, A.M. Varicella-zoster virus infection of human dorsal root ganglia in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 6490–6495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouwendijk, W.J.D.; Mahalingam, R.; de Swart, R.L.; Haagmans, B.L.; van Amerongen, G.; Getu, S.; Gilden, D.; Osterhaus, A.D.M.E.; Verjans, G.M.G.M. T-Cell Tropism of Simian Varicella Virus during Primary Infection. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Croen, K.D.; Ostrove, J.M.; Dragovic, L.J.; Straus, S.E. Patterns of gene expression and sites of latency in human nerve ganglia are different for varicella-zoster and herpes simplex viruses. Proc. Natl. Acad. Sci. USA 1988, 85, 9773–9777. [Google Scholar] [CrossRef] [PubMed]
- Steain, M.; Slobedman, B.; Abendroth, A. Experimental models to study varicella-zoster virus infection of neurons. Curr. Top. Microbiol. Immunol. 2010. [Google Scholar] [CrossRef]
- Plotkin, S.A.; Stein, S.; Snyder, M.; Immesoete, P. Attempts to recover varicella virus from ganglia. Ann. Neurol. 1977, 2, 249. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Lebenthal-Loinger, I.; Yang, I.H.; Kinchington, P.R.; Goldstein, R.S. An In Vitro Model of Latency and Reactivation of Varicella Zoster Virus in Human Stem Cell-Derived Neurons. PLoS Pathog. 2015, 11, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerboni, L.; Sobel, R.A.; Ramachandran, V.; Rajamani, J.; Ruyechan, W.; Abendroth, A.; Arvin, A. Expression of varicella-zoster virus immediate-early regulatory protein IE63 in neurons of latently infected human sensory ganglia. J. Virol. 2010, 84, 3421–3430. [Google Scholar] [CrossRef] [PubMed]
- Grinfeld, E.; Kennedy, P.G.E. Translation of varicella-zoster virus genes during human ganglionic latency. Virus Genes 2004, 29, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, R.; Wellish, M.; Cohrs, R.; Debrus, S.; Piette, J.; Rentier, B.; Gilden, D.H. Expression of protein encoded by varicella-zoster virus open reading frame 63 in latently infected human ganglionic neurons. Proc. Natl. Acad. Sci. USA 1996, 93, 2122–2124. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.G.; Grinfeld, E.; Bell, J.E. Varicella-zoster virus gene expression in latently infected and explanted human ganglia. J. Virol. 2000, 74, 11893–11898. [Google Scholar] [CrossRef] [PubMed]
- Cohrs, R.J.; Gilden, D.H.; Kinchington, P.R.; Grinfeld, E.; Kennedy, P.G.E. Varicella-zoster virus gene 66 transcription and translation in latently infected human Ganglia. J. Virol. 2003, 77, 6660–6665. [Google Scholar] [CrossRef] [PubMed]
- Zerboni, L.; Sobel, R.A.; Lai, M.; Triglia, R.; Steain, M.; Abendroth, A.; Arvin, A. Apparent expression of varicella-zoster virus proteins in latency resulting from reactivity of murine and rabbit antibodies with human blood group a determinants in sensory neurons. J. Virol. 2012, 86, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Ouwendijk, W.J.D.; Flowerdew, S.E.; Wick, D.; Horn, A.K.E.; Sinicina, I.; Strupp, M.; Osterhaus, A.D.M.E.; Verjans, G.M.G.M.; Hüfner, K. Immunohistochemical detection of intra-neuronal VZV proteins in snap-frozen human ganglia is confounded by antibodies directed against blood group A1-associated antigens. J. Neurovirol. 2012, 18, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.C.; Mohr, I. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol. 2012, 20, 604–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouwendijk, W.J.D.; Choe, A.; Nagel, M.A.; Gilden, D.; Osterhaus, A.D.M.E.; Cohrs, R.J.; Verjans, G.M.G.M. Restricted varicella-zoster virus transcription in human trigeminal ganglia obtained soon after death. J. Virol. 2012, 86, 10203–10206. [Google Scholar] [CrossRef] [PubMed]
- Hafezi, W.; Lorentzen, E.U.; Eing, B.R.; Muller, M.; King, N.J.C.; Klupp, B.; Mettenleiter, T.C.; Kuhn, J.E. Entry of herpes simplex virus type 1 (HSV-1) into the distal axons of trigeminal neurons favors the onset of nonproductive, silent infection. PLoS Pathog. 2012, 8, e1002679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoll, M.P.; Proenca, J.T.; Efstathiou, S. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 2012, 36, 684–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gary, L.; Gilden, D.H.; Cohrs, R.J. Epigenetic regulation of varicella-zoster virus open reading frames 62 and 63 in latently infected human trigeminal ganglia. J. Virol. 2006, 80, 4921–4926. [Google Scholar] [CrossRef] [PubMed]
- Amelio, A.L.; McAnany, P.K.; Bloom, D.C. A chromatin insulator-like element in the herpes simplex virus type 1 latency-associated transcript region binds CCCTC-binding factor and displays enhancer-blocking and silencing activities. J. Virol. 2006, 80, 2358–2368. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Davis, K.A.; Traina-Dorge, V.; Gray, W.L. Simian varicella virus expresses a latency-associated transcript that is antisense to open reading frame 61 (ICP0) mRNA in neural ganglia of latently infected monkeys. J. Virol. 2007, 81, 8149–8156. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, I.; Barron, A.; Wellish, M.; Engelmann, F.; Legasse, A.; Planer, S.; Gilden, D.; Nikolich-Zugich, J.; Mahalingam, R. Simian varicella virus infection of rhesus macaques recapitulates essential features of varicella zoster virus infection in humans. PLoS Pathog. 2009, 5, e1000657. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.G.; Wagner, E.K.; Devi-Rao, G.B.; Cook, M.L.; Feldman, L.T. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 1987, 235, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaber, T.; Workman, A.; Jones, C. Small Noncoding RNAs Encoded within the Bovine Herpesvirus 1 Latency-Related Gene Can Reduce Steady-State Levels of Infected Cell Protein 0 (bICP0). J. Virol. 2010, 84, 6297–6307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahjoub, N.; Dhorne-Pollet, S.; Fuchs, W.; Endale Ahanda, M.-L.; Lange, E.; Klupp, B.; Arya, A.; Loveland, J.E.; Lefevre, F.; Mettenleiter, T.C.; et al. A 2.5-Kilobase Deletion Containing a Cluster of Nine MicroRNAs in the Latency-Associated-Transcript Locus of the Pseudorabies Virus Affects the Host Response of Porcine Trigeminal Ganglia during Established Latency. J. Virol. 2015, 89, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Roizman, B.; Zhou, G. The 3 facets of regulation of herpes simplex virus gene expression: A. critical inquiry. Virology 2015, 479–480, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Scherba, G. Expression of the pseudorabies virus latency-associated transcript gene during productive infection of cultured cells. J. Virol. 1999, 73, 9781–9788. [Google Scholar] [PubMed]
- Devireddy, L.R.; Jones, C. Alternative splicing of the latency-related transcript of bovine herpesvirus 1 yields RNAs containing unique open reading frames. J. Virol. 1998, 72, 7294–7301. [Google Scholar] [PubMed]
- Sinani, D.; Frizzo da Silva, L.; Jones, C. A Bovine Herpesvirus 1 Protein Expressed in Latently Infected Neurons (ORF2) Promotes Neurite Sprouting in the Presence of Activated Notch1 or Notch3. J. Virol. 2013, 87, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Maillet, S.; Naas, T.; Crepin, S.; Roque-Afonso, A.-M.; Lafay, F.; Efstathiou, S.; Labetoulle, M. Herpes Simplex Virus Type 1 Latently Infected Neurons Differentially Express Latency-Associated and ICP0 Transcripts. J. Virol. 2006, 80, 9310–9321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-H.; Lee, L.Y.; Garber, D.A.; Schaffer, P.A.; Knipe, D.M.; Coen, D.M. Neither LAT nor open reading frame P mutations increase expression of spliced or intron-containing ICP0 transcripts in mouse ganglia latently infected with herpes simplex virus. J. Virol. 2002, 76, 4764–4772. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Selariu, A.; Warden, C.; Huang, G.; Huang, Y.; Zaccheus, O.; Cheng, T.; Xia, N.; Zhu, H. Genome-wide mutagenesis reveals that ORF7 is a novel VZV skin-tropic factor. PLoS Pathog. 2010, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jackers, P.; Defechereux, P.; Baudoux, L.; Lambert, C.; Massaer, M.; Merville-Louis, M.P.; Rentier, B.; Piette, J. Characterization of regulatory functions of the varicella-zoster virus gene 63-encoded protein. J. Virol. 1992, 66, 3899–3903. [Google Scholar] [PubMed]
- Verweij, M.C.; Wellish, M.; Whitmer, T.; Malouli, D.; Lapel, M.; Jonjić, S.; Haas, J.G.; DeFilippis, V.R.; Mahalingam, R.; Früh, K. Varicella Viruses Inhibit Interferon-Stimulated JAK-STAT Signaling through Multiple Mechanisms. PLoS Pathog. 2015, 11, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Ambagala, A.P.N.; Cohen, J.I. Varicella-Zoster virus IE63, a major viral latency protein, is required to inhibit the alpha interferon-induced antiviral response. J. Virol. 2007, 81, 7844–7851. [Google Scholar] [CrossRef] [PubMed]
- Gerada, C.; Steain, M.; McSharry, B.P.; Slobedman, B.; Abendroth, A. VZV ORF63 protects human neuronal and keratinocyte cell lines from apoptosis and changes its localization upon apoptosis induction. J. Virol. 2018, JVI.00338-18. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.S.; Kyratsous, C.A.; Wan, S.; Silverstein, S. Nuclear import of the varicella-zoster virus latency-associated protein ORF63 in primary neurons requires expression of the lytic protein ORF61 and occurs in a proteasome-dependent manner. J. Virol. 2008, 82, 8673–8686. [Google Scholar] [CrossRef] [PubMed]
- van Velzen, M.; Ouwendijk, W.J.D.; Selke, S.; Pas, S.D.; van Loenen, F.B.; Osterhaus, A.D.M.E.; Wald, A.; Verjans, G.M.G.M. Longitudinal study on oral shedding of herpes simplex virus 1 and varicella-zoster virus in individuals infected with HIV. J. Med. Virol. 2013, 85, 1669–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esiri, M.M.; Tomlinson, A.H. Herpes zoster. Demonstration of virus in trigeminal nerve and ganglion by immunofluorescence and electron microscopy. J. Neurol. Sci. 1972, 15, 35–48. [Google Scholar] [CrossRef]
- Nagashima, K.; Nakazawa, M.; Endo, H. Pathology of the human spinal ganglia in varicella-zoster virus infection. Acta Neuropathol. 1975, 33, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Joesoef, R.M.; Bialek, S.; Wang, C.; Harpaz, R. Association of physical trauma with risk of herpes zoster among medicare beneficiaries in the United States. J. Infect. Dis. 2013, 207, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, E.K.; Brown, C.; Stambough, J.L. Herpes zoster-varicella complicating anterior thoracic surgery: 2 Case reports. J. Spinal Disord. Tech. 2006, 19, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Simms, H.N.; Dunn, L.T. Herpes zoster of the trigeminal nerve following microvascular decompression. Br. J. Neurosurg. 2006, 20, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Delcroix, J.D.; Valletta, J.S.; Wu, C.; Hunt, S.J.; Kowal, A.S.; Mobley, W.C. NGF signaling in sensory neurons: Evidence that early endosomes carry NGF retrograde signals. Neuron 2003, 39, 69–84. [Google Scholar] [CrossRef]
- Cohrs, R.J.; Badani, H.; Baird, N.L.; White, T.M.; Sanford, B.; Gilden, D. Induction of varicella zoster virus DNA replication in dissociated human trigeminal ganglia. J. Neurovirol. 2017, 23, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Kurapati, S.; Sadaoka, T.; Rajbhandari, L.; Shukla, P.; Ali, M.A.; Kim, Y.J.; Lee, G.; Cohen, J.I.; Venkatesan, A. Role of the JNK pathway in varicella-zoster virus lytic infection and reactivation. J. Virol. 2017, 91, e00640-17. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Vogel, J.L.; Narayanan, A.; Peng, H.; Kristie, T.M. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat. Med. 2009, 15, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal Stress Pathway Mediating a Histone Methyl/Phospho Switch Is Required for Herpes Simplex Virus Reactivation. Cell Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Zerboni, L.; Arvin, A. Neuronal Subtype and Satellite Cell Tropism Are Determinants of Varicella-Zoster Virus Virulence in Human Dorsal Root Ganglia Xenografts In Vivo. PLoS Pathog. 2015, 11, e1004989. [Google Scholar] [CrossRef] [PubMed]
- Enquist, L.W.; Leib, D.A. Intrinsic and Innate Defenses of Neurons: Détente with the Herpesviruses. J. Virol. 2017, 91, JVI.01200-16. [Google Scholar] [CrossRef] [PubMed]
- Hanani, M. Satellite glial cells in sensory ganglia: From form to function. Brain Res. Rev. 2005, 48, 457–476. [Google Scholar] [CrossRef] [PubMed]
- van Velzen, M.; Laman, J.D.; Kleinjan, A.; Poot, A.; Osterhaus, A.D.M.E.; Verjans, G.M.G.M. Neuron-interacting satellite glial cells in human trigeminal ganglia have an APC phenotype. J. Immunol. 2009, 183, 2456–2461. [Google Scholar] [CrossRef] [PubMed]
- Mitterreiter, J.G.; Ouwendijk, W.J.D.; van Velzen, M.; van Nierop, G.P.; Osterhaus, A.D.M.E.; Verjans, G.M.G.M. Satellite glial cells in human trigeminal ganglia have a broad expression of functional Toll-like receptors. Eur. J. Immunol. 2017, 47, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Mori, I.; Goshima, F.; Koshizuka, T.; Imai, Y.; Kohsaka, S.; Koide, N.; Sugiyama, T.; Yoshida, T.; Yokochi, T.; Kimura, Y.; et al. Iba1-expressing microglia respond to herpes simplex virus infection in the mouse trigeminal ganglion. Mol. Brain Res. 2003, 120, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Ouwendijk, W.J.D.; Getu, S.; Mahalingam, R.; Gilden, D.; Osterhaus, A.D.M.E.; Verjans, G.M.G.M. Characterization of the immune response in ganglia after primary simian varicella virus infection. J. Neurovirol. 2016, 22, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.E. Selective decline in cellular immune response to varicella-zoster in the elderly. Neurology 1980, 30, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.J.; Smith, J.G.; Kaufhold, R.M.; Barber, D.; Hayward, A.R.; Chan, C.Y.; Chan, I.S.F.; Li, D.J.J.; Wang, W.; Keller, P.M.; et al. Decline in varicella-zoster virus (VZV)-specific cell-mediated immunity with increasing age and boosting with a high-dose VZV vaccine. J. Infect. Dis. 2003, 188, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Amsen, D.; van Gisbergen, K.P.J.M.; Hombrink, P.; van Lier, R.A.W. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat. Immunol. 2018, 19, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, T.; Palendira, U.; Tscharke, D.C.; Bedoui, S. Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol. Rev. 2018, 283, 54–76. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Stock, A.T.; Ma, J.Z.; Jones, C.M.; Kent, S.J.; Mueller, S.N.; Heath, W.R.; Carbone, F.R.; Gebhardt, T. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc. Natl. Acad. Sci. USA 2012, 109, 7037–7042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knickelbein, J.E.; Khanna, K.M.; Yee, M.B.; Baty, C.J.; Kinchington, P.R.; Hendricks, R.L. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 2008, 322, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.M.; Bonneau, R.H.; Kinchington, P.R.; Hendricks, R.L. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity 2003, 18, 593–603. [Google Scholar] [CrossRef]
- van Velzen, M.; Jing, L.; Osterhaus, A.D.M.E.; Sette, A.; Koelle, D.M.; Verjans, G.M.G.M. Local CD4 and CD8 T-cell reactivity to HSV-1 antigens documents broad viral protein expression and immune competence in latently infected human trigeminal ganglia. PLoS Pathog. 2013, 9, e1003547. [Google Scholar] [CrossRef] [PubMed]
- Vukmanovic-Stejic, M.; Sandhu, D.; Seidel, J.A.; Patel, N.; Sobande, T.O.; Agius, E.; Jackson, S.E.; Fuentes-Duculan, J.; Suarez-Farinas, M.; Mabbott, N.A.; et al. The Characterization of Varicella Zoster Virus Specific T Cells In Skin and Blood during Ageing Europe PMC Funders Group. J. Investig. Dermatol. 2015, 135, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Steain, M.; Sutherland, J.P.; Rodriguez, M.; Cunningham, A.L.; Slobedman, B.; Abendroth, A. Analysis of T cell responses during active varicella-zoster virus reactivation in human ganglia. J. Virol. 2014, 88, 2704–2716. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, K.; Steain, M.; Cunningham, A.L.; Rodriguez, M.; Blumbergs, P.; Slobedman, B.; Abendroth, A. Characterization of the host immune response in human Ganglia after herpes zoster. J. Virol. 2010, 84, 8861–8870. [Google Scholar] [CrossRef] [PubMed]
- Verjans, G.M.G.M.; Hintzen, R.Q.; van Dun, J.M.; Poot, A.; Milikan, J.C.; Laman, J.D.; Langerak, A.W.; Kinchington, P.R.; Osterhaus, A.D.M.E. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc. Natl. Acad. Sci. USA 2007, 104, 3496–3501. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, T.J.; McCarthy, M.; Cohrs, R.J.; Kaufer, B.B. 3D tissue-like assemblies: A novel approach to investigate virus-cell interactions. Methods 2015, 90, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Sadaoka, T.; Schwartz, C.L.; Rajbhandari, L.; Venkatesan, A.; Cohen, J.I. Human Embryonic Stem Cell-Derived Neurons Are Highly Permissive for Varicella-Zoster Virus Lytic Infection. J. Virol. 2018, 92, JVI-01108. [Google Scholar] [CrossRef] [PubMed]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lönnerberg, P.; Lou, D.; Hjerling-Leffler, J.; Haeggström, J.; Kharchenko, O.; Kharchenko, P.V.; et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015, 18, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Flowerdew, S.E.; Wick, D.; Himmelein, S.; Horn, A.K.E.; Sinicina, I.; Strupp, M.; Brandt, T.; Theil, D.; Hüfner, K. Characterization of Neuronal Populations in the Human Trigeminal Ganglion and Their Association with Latent Herpes Simplex Virus-1 Infection. PLoS ONE 2013, 8, e83603. [Google Scholar] [CrossRef] [PubMed]
- Bertke, A.S.; Patel, A.; Imai, Y.; Apakupakul, K.; Margolis, T.P.; Krause, P.R. Latency-Associated Transcript (LAT) Exon 1 Controls Herpes Simplex Virus Species-Specific Phenotypes: Reactivation in the Guinea Pig Genital Model and Neuron Subtype-Specific Latent Expression of LAT. J. Virol. 2009, 83, 10007–10015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Mandarino, A.; Chao, M.V.; Mohr, I.; Wilson, A.C. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog. 2012, 8, e1002540. [Google Scholar] [CrossRef] [PubMed]
- Vukmanovic-Stejic, M.; Sandhu, D.; Sobande, T.O.; Agius, E.; Lacy, K.E.; Riddell, N.; Montez, S.; Dintwe, O.B.; Scriba, T.J.; Breuer, J.; et al. Varicella Zoster Specific CD4 + Foxp3 + T Cells Accumulate after Cutaneous Antigen Challenge in Humans. J. Immunol. 2013, 1, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Linderman, J.A.; Kobayashi, M.; Rayannavar, V.; Fak, J.J.; Darnell, R.B.; Chao, M.V.; Wilson, A.C.; Mohr, I. Immune Escape via a Transient Gene Expression Program Enables Productive Replication of a Latent Pathogen. Cell Rep. 2017, 18, 1312–1323. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Depledge, D.P.; Sadaoka, T.; Ouwendijk, W.J.D. Molecular Aspects of Varicella-Zoster Virus Latency. Viruses 2018, 10, 349. https://doi.org/10.3390/v10070349
Depledge DP, Sadaoka T, Ouwendijk WJD. Molecular Aspects of Varicella-Zoster Virus Latency. Viruses. 2018; 10(7):349. https://doi.org/10.3390/v10070349
Chicago/Turabian StyleDepledge, Daniel P., Tomohiko Sadaoka, and Werner J. D. Ouwendijk. 2018. "Molecular Aspects of Varicella-Zoster Virus Latency" Viruses 10, no. 7: 349. https://doi.org/10.3390/v10070349
APA StyleDepledge, D. P., Sadaoka, T., & Ouwendijk, W. J. D. (2018). Molecular Aspects of Varicella-Zoster Virus Latency. Viruses, 10(7), 349. https://doi.org/10.3390/v10070349