Identification of Novel HIV-1 Latency-Reversing Agents from a Library of Marine Natural Products

 , , ,
, , ,
Abstract
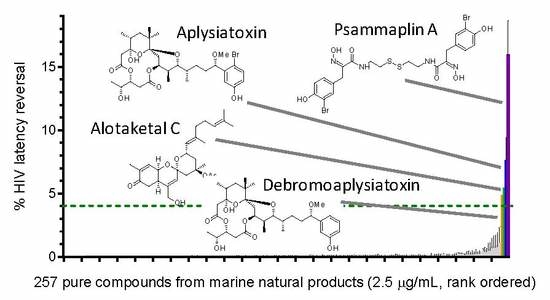
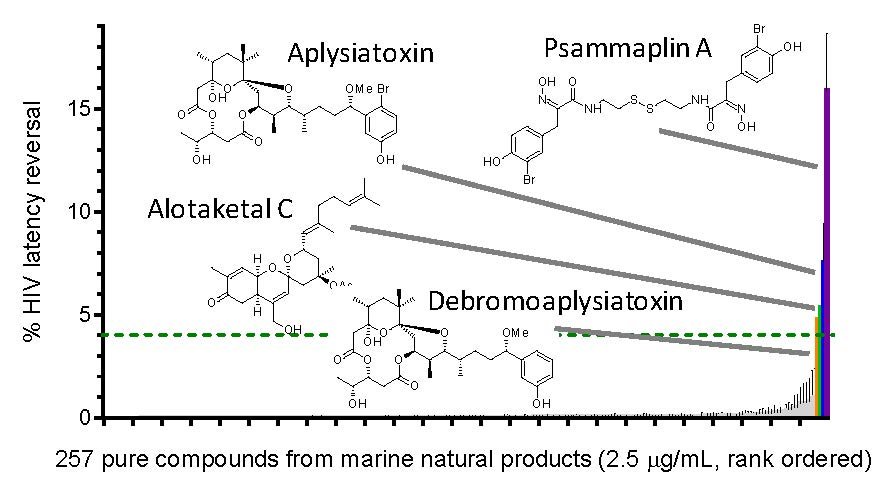
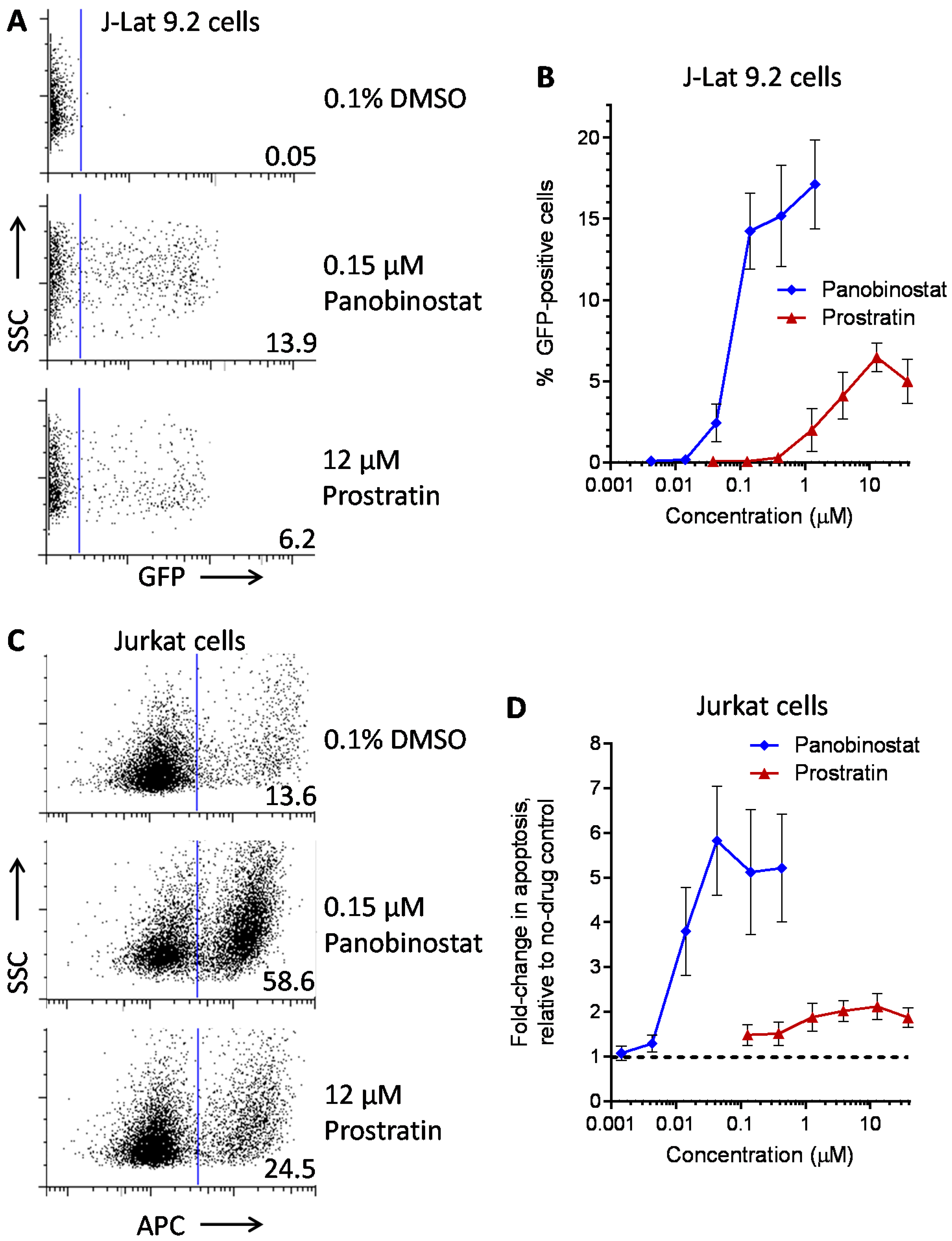
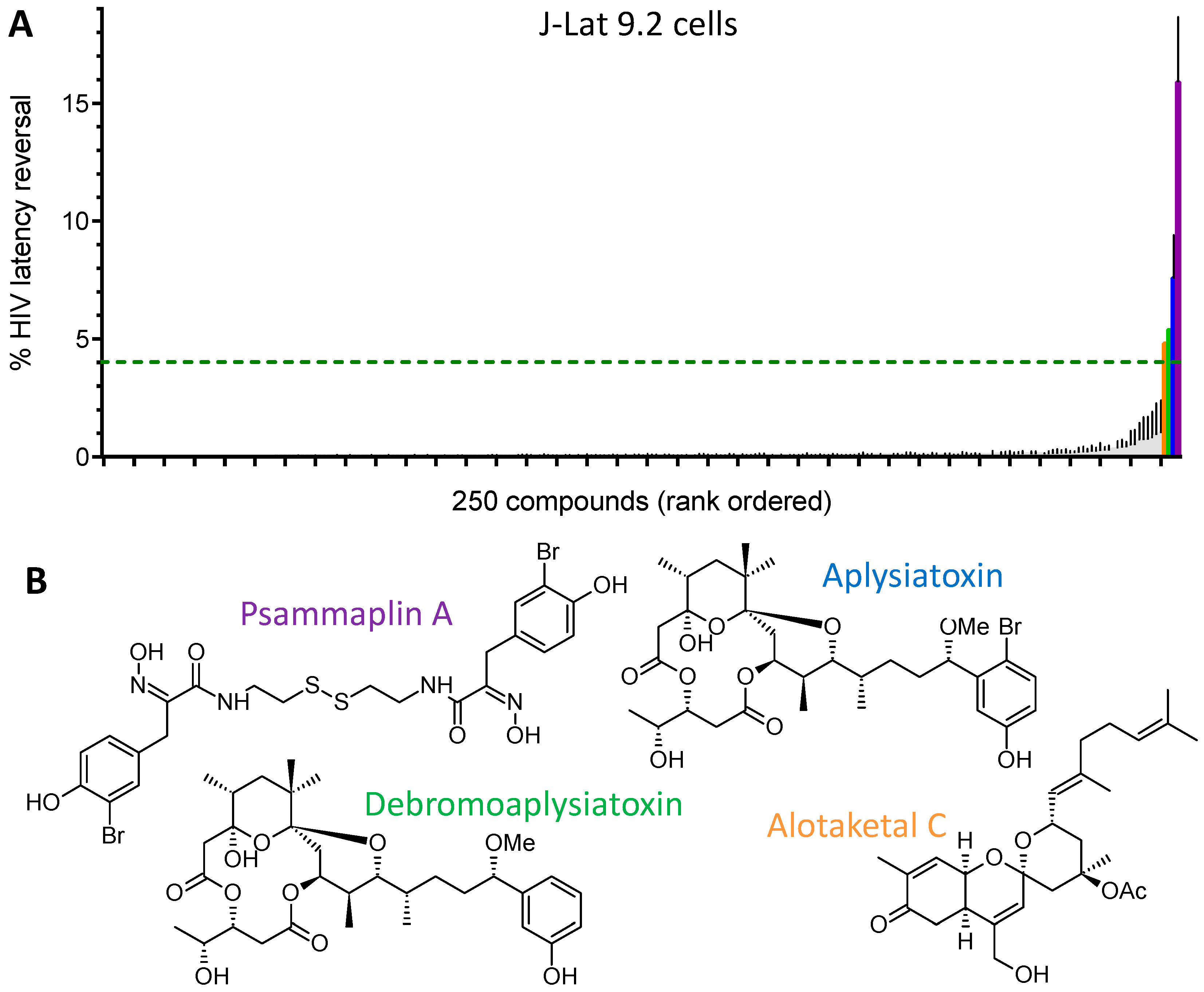
:
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Molecular Mechanisms of HIV latency. J. Clin. Investig. 2016, 126, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G. HIV: Shock and kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Margolis, D.M. Emerging strategies to deplete the HIV reservoir. Curr. Opin. Infect. Dis. 2014, 27, 29–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spivak, A.M.; Planelles, V. Novel latency reversal agents for HIV-1 cure. Annu. Rev. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.A.; Lewin, S.R. Shocking HIV out of hiding: Where are with with clinical trials of latency reversing agents? Curr. Opin. HIV AIDS 2016, 11, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Reuse, S.; Calao, M.; Kabeya, K.; Guiguen, A.; Gatot, J.S.; Quivy, V.; Vanhulle, C.; Lamine, A.; Vaira, D.; Demonte, D.; et al. Synertistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: Implications for treatment of latent infection. PLoS ONE 2009, 4, e6093. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.; de Vinuesa, A.G.; Sanchez-Duffhues, G.; Marquez, N.; Bellido, M.L.; Muñoz-Fernández, M.Á.; Moreno, S.; Castor, T.P.; Calzado, M.A.; Muñoz, E. Bryostatin-1 synergizes with histone deacteylase inhibitors to reactivate HIV-1 from latency. Curr. HIV Res. 2010, 8, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.; Martin, A.R.; Hill, A.L.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J. Clin. Investig. 2015, 125, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Mendes, E.A.; Kaiser, P.; Wong, D.P.; Tang, Y.; Cai, I.; Fenton, A.; Melcher, G.P.; Hildreth, J.E.; Thompson, G.R.; et al. Synergistic reactivation of latent HIV expression by ingenol-3-angelate, PEP005, targeted NF-κB signaling in combination with JQ1 induced p-TEFB activation. PLoS Pathog. 2015, 11, e1005066. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Kula, A.; Bouchat, S.; Fujinaga, K.; Corazza, F.; Ait-Ammar, A.; Delacourt, N.; Melard, A.; Kabeya, K.; Vanhulle, C.; et al. An in-depth comparison of latency-reversing agent combinations in various in vitro and ex vivo HIV-1 latency models identified bryostatin-1+ JQ1 and ingenol-B+ JQ1 to potently reactivate viral gene expression. PLoS Pathog. 2015, 11, e1005063. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Bonet, M.; Clemente, M.I.; Serramía, M.J.; Muñoz, E.; Moreno, S.; Muñoz-Fernández, M.Á. Synergistic activation of latent HIV-1 expression by novel histone deacetylase inhibitors and bryostatin-1. Sci. Rep. 2015, 5, 16445. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tietjen, I.; Chen, M.; Williams, D.E.; Daoust, J.; Brockman, M.A.; Andersen, R.J. Sesterterpenoids isolated from the sponge Phorbas sp. activate latent HIV-1 provirus expression. J. Org. Chem. 2016, 81, 11324–11334. [Google Scholar] [CrossRef] [PubMed]
- Tietjen, I.; Ngwenya, B.N.; Fotso, G.; Williams, D.E.; Simonambango, S.; Ngadjui, B.T.; Andersen, R.J.; Brockman, M.A.; Brumme, Z.L.; Andrae-Marobela, K. The Croton megalobotrys Müll Arg. traditional medicine in HIV/AIDS management: Documentation of patient use, in vitro activation of latent HIV-1 provirus, and isolation of active phorbol esters. J. Ethnopharmacol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, J.; Yang, B.; Lin, X.; Yang, X.W.; Liu, Y. Marine natural products with anti-HIV activities in the last decade. Curr. Med. Chem. 2013, 20, 953–973. [Google Scholar] [PubMed]
- Cary, D.C.; Peterlin, D.M. Natural products and HIV/AIDS. AIDS Res. Hum. Retrovir. 2018, 34, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.C.; Xing, S.; Shan, L.; O’Connell, K.; Dinoso, J.; Shen, A.; Zhou, Y.; Shrum, C.K.; Han, Y.; Liu, J.O.; et al. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J. Clin. Investig. 2009, 119, 3473–3485. [Google Scholar] [CrossRef] [PubMed]
- Doyon, G.; Sobolewski, M.D.; Huber, K.; McMahon, D.; Mellors, J.W.; Sluis-Cremer, N. Discovery of a small molecule agonist of phosphatidylinositol 3-kinase p110α that reactivates latent HIV-1. PLoS ONE 2014, 9, e84964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daoust, J.; Chen, M.; Wang, M.; Williams, D.E.; Garcia Chavez, M.A.; Yang, Y.A.; Merchant, C.E.; Fontana, A.; Kieffer, T.J.; Andersen, R.J. Sesterterpenoids isolated from a northeaster Pacific Phorbas sp. J. Org. Chem. 2013, 78, 8267–8273. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Carr, G.; Zhang, Y.; Williams, D.E.; Amlani, A.; Bottriell, H.; Mui, A.L.-F.; Andersen, R.J. Turnagainolides A and B, cyclic depsipeptides produced in culture by a Bacillus sp.: Isolation, structure elucidation, and synthesis. J. Nat. Prod. 2011, 74, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.E.; Dalisay, D.S.; Patrick, B.O.; Matainaho, T.; Andrusiak, K.; Deshpande, R.; Myers, C.L.; Piotrowski, J.S.; Boone, C.; Yoshida, M.; et al. Padanamides A and B, highly modified linear tetrapeptides produced in culture by a Streptomyces sp. isolated from marine sediment. Org. Lett. 2011, 13, 3936–3939. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.E.; Steinø, A.; de Voogd, N.J.; Mauk, A.G.; Andersen, R.J. Halicloic acids A and B isolated from the marine sponge Haliclona sp. collected in the Phillipines inhibit indoleamine 2,3-dioxygenase (IDO). J. Nat. Prod. 2012, 75, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.E.; Dalisay, D.S.; Li, F.; Amphlett, J.; Maneerat, W.; Matainaho, T.; Yu, W.; Brown, P.J.; Arrowsmith, C.H.; Vedadi, M.; et al. Nahuoic acid A produced by a Streptomyces sp. isolated from a marine sediment is a selective SAM-competitive inhibitor of the histone methyltransferase SETD8. Org. Lett. 2013, 15, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Tietjen, I.; Williams, D.E.; Read, S.; Kuang, X.T.; Mwimanzi, P.; Wilhelm, E.; Markle, T.; Kinloch, N.N.; Naphen, C.N.; Tenney, K.; et al. Inhibition of NF-κB-dependent HIV-1 replication by the marine natural product bengamide A. Antivir. Res. 2018, 152, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spina, C.A.; Anderson, J.; Archin, N.M.; Bosque, A.; Chan, J.; Famiglietti, M.; Greene, W.C.; Kashuba, A.; Lewin, S.R.; Margolis, D.M.; et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell models systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 2013, 9, e1003834. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, P.; Barreto, K.; Bernhard, W.; Lomness, A.; Nonson, N.; Pfeifer, T.A.; Harrigan, P.R.; Sadowski, I. Compounds producing an effective combinatorial regimen for disruption of HIV-1 latency. EMBO Mol. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Sim, C.J.; Lee, C.O. Cytotoxic compounds from a two-sponge association. J. Nat. Prod. 1995, 58, 1722–1726. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Shin, J.; Kwon, H.J. Psammaplin A is a natural prodrug that inhibits class I histone deacetylase. Exp. Mol. Med. 2007, 39, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baud, M.G.; Leiser, T.; Haus, P.; Samlal, S.; Wong, A.C.; Wood, R.J.; Petrucci, V.; Gunaratnam, M.; Hughes, S.M.; Buluwela, L.; et al. Defining the mechanism of action and enzymatic selectivity of psammaplin A against its epigenetic targets. J. Med. Chem. 2012, 55, 1731–1750. [Google Scholar] [CrossRef] [PubMed]
- Nagai, H.; Yasumoto, T.; Hokama, Y. Aplysiatoxin and debromoaplysiatoxin as the causative agents of a red alga Gracilaria coronopifolia poisoning in Hawaii. Toxicon 1996, 34, 753–761. [Google Scholar] [CrossRef]
- Arcoleo, J.P.; Weinstein, I.B. Activation of protein kinase C by tumor promoting phorbol esters, teleocidin and aplysiatoxin in the absence of added calcium. Carcinogenesis 1985, 6, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Kishi, Y.; Pajares, M.A.; Rando, R.R. Structural basis of protein kinase C activation by tumor promoters. Proc. Natl. Acad. Sci. USA 1989, 86, 9672–9676. [Google Scholar] [CrossRef] [PubMed]
- Micheva-Viteva, S.; Kobayashi, Y.; Edelstein, L.C.; Pacchia, A.L.; Lee, H.L.R.; Graci, J.D.; Breslin, J.; Phelan, B.D.; Miller, L.K.; Colacino, J.M.; et al. High-throughput screening uncovers a compound that activates latent HIV-1 and acts cooperatively with a histone deacetylase (HDAC) inhibitor. J. Biol. Chem. 2011, 286, 21083–21091. [Google Scholar] [CrossRef] [PubMed]
- Gallastegui, E.; Marshall, B.; Vidal, D.; Sanchez-Duffhues, G.; Collado, J.A.; Alvarez-Fernández, C.; Luque, N.; Terme, J.M.; Gatell, J.M.; Sánchez-Palomino, S.; et al. Combination of biological screening in a cellular model of viral latency and virtual screening identifies novel compounds that reactivate HIV-1. J. Virol. 2012, 86, 3795–3808. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Xing, S.; Yang, H.-C.; Zhang, H.; Margolick, J.B.; Siliciano, R.F. Unique characteristics of histone deacetylase inhibitors in reactivation of latent HIV-1 in Bcl-2-transduced primary resting CD4+ T cells. J. Antimicrob. Chemother. 2014, 69, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Beans, E.J.; Fournogerakis, D.; Gauntlett, C.; Heumann, L.V.; Kramer, R.; Marsden, M.D.; Murray, D.; Chun, T.-W.; Zack, J.A.; Wender, P.A. Highly potent, synthetically accessible prostratin analogs induce latent HIV expression in vitro and ex vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 11698–11703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romani, B.; Jamil, R.K.; Hamidi-Fard, M.; Rahimi, P.; Momen, S.B.; Aghasadeghi, M.R.; Allahbakhshi, E. HIV-1 Vpr reactivates latent HIV-1 provirus by inducing depletion of class I HDACs on chromatin. Sci. Rep. 2016, 6, 31924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbi, M.F.; Al-Harthi, L.; Saifuddin, M.; Roebuck, K.A. The cAMP-dependent protein kinase A and protein kinase C-beta pathways synergistically interact to activate HIV-1 transcription in latently infected cells of monocyte/macrophage lineage. Virology 1998, 245, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Kim, K.C.; Son, J.; Shin, Y.; Yoon, C.H.; Kang, C.; Choi, B.S. Synergistic reactivation of latent HIV-1 provirus by PKA activator dibutryl-cAMP in combination with an HDAC inhibitor. Virus Res. 2017, 227, 1–5. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
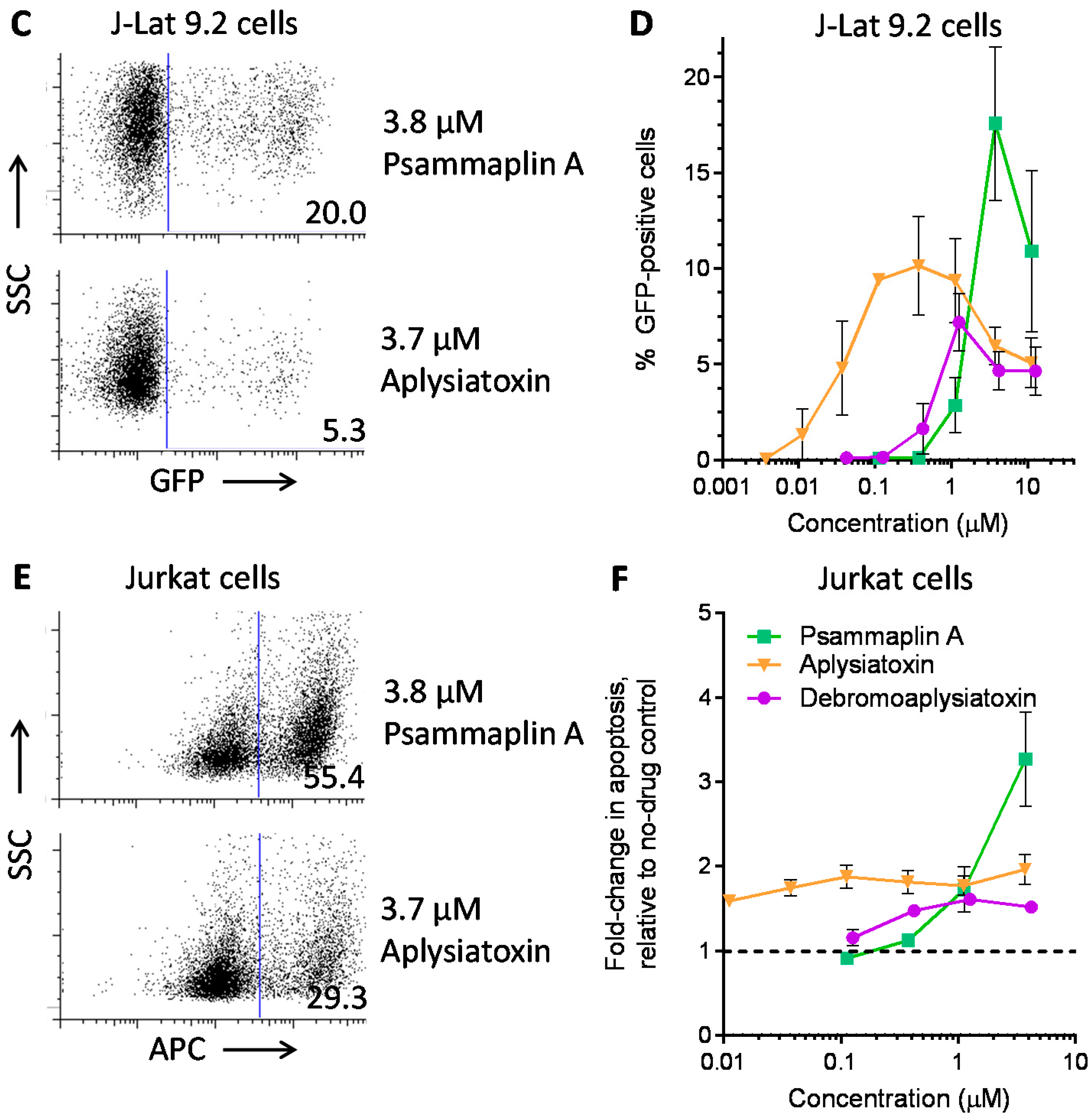
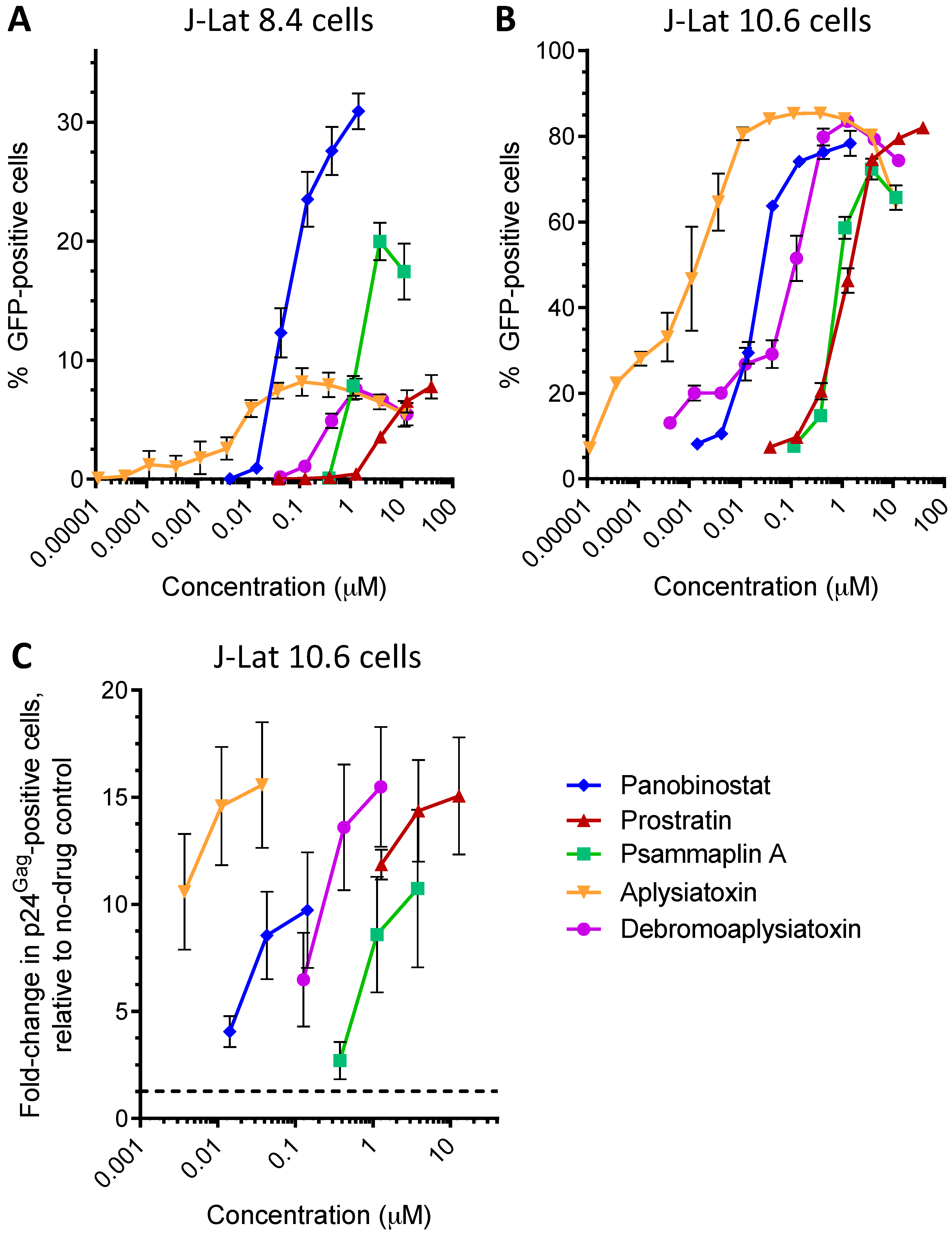
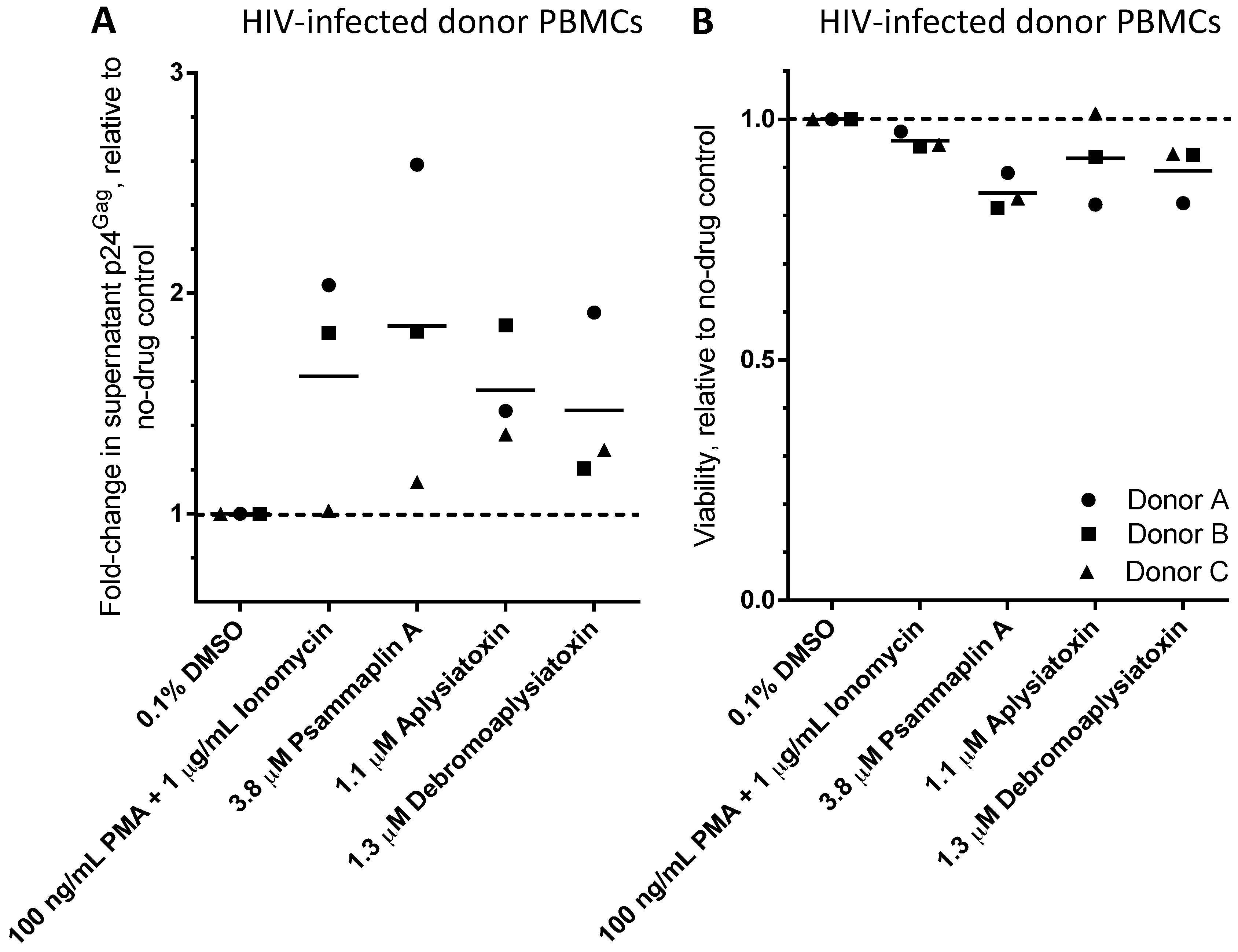
| LRA | EC50 (µM; mean ± s.e.m.) | Mechanism of Action | Ref. | ||
|---|---|---|---|---|---|
| J-Lat 9.2 | J-Lat 8.4 | J-Lat 10.6 | |||
| Panobinostat | 0.10 ± 0.02 | 0.073 ± 0.010 | 0.041 ± 0.003 | HDACi | [5,6] |
| Prostratin | 7.1 ± 2.8 | 10 ± 1 | 1.8 ± 0.4 | PKC activator | [5,6] |
| Psammaplin A | 1.9 ± 0.3 | 1.5 ± 0.1 | 1.5 ± 0.1 | HDACi | [29,30,31] |
| Aplysiatoxin | 0.045 ± 0.021 | 0.011 ± 0.003 | 0.0033 ± 0.0012 | PKC activator | [32,33,34] |
| Debromoaplysiatoxin | 0.92 ± 0.14 | 0.52 ± 0.02 | 0.081 ± 0.029 | PKC activator | [32,34] |
| Alotaketal C | 1.3 ± 0.2 | n.d. | n.d. | PKC activator | [14] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richard, K.; Williams, D.E.; De Silva, E.D.; Brockman, M.A.; Brumme, Z.L.; Andersen, R.J.; Tietjen, I. Identification of Novel HIV-1 Latency-Reversing Agents from a Library of Marine Natural Products. Viruses 2018, 10, 348. https://doi.org/10.3390/v10070348
Richard K, Williams DE, De Silva ED, Brockman MA, Brumme ZL, Andersen RJ, Tietjen I. Identification of Novel HIV-1 Latency-Reversing Agents from a Library of Marine Natural Products. Viruses. 2018; 10(7):348. https://doi.org/10.3390/v10070348
Chicago/Turabian StyleRichard, Khumoekae, David E. Williams, E. Dilip De Silva, Mark A. Brockman, Zabrina L. Brumme, Raymond J. Andersen, and Ian Tietjen. 2018. "Identification of Novel HIV-1 Latency-Reversing Agents from a Library of Marine Natural Products" Viruses 10, no. 7: 348. https://doi.org/10.3390/v10070348
APA StyleRichard, K., Williams, D. E., De Silva, E. D., Brockman, M. A., Brumme, Z. L., Andersen, R. J., & Tietjen, I. (2018). Identification of Novel HIV-1 Latency-Reversing Agents from a Library of Marine Natural Products. Viruses, 10(7), 348. https://doi.org/10.3390/v10070348