Mimiviridae: An Expanding Family of Highly Diverse Large dsDNA Viruses Infecting a Wide Phylogenetic Range of Aquatic Eukaryotes



Abstract
:1. Introduction
2. Results
2.1. Pionnering Work on Large Aquatic Viruses
2.2. From Acanthamoeba Polyphaga Mimivirus (APMV) to the First Marine Mimiviridae
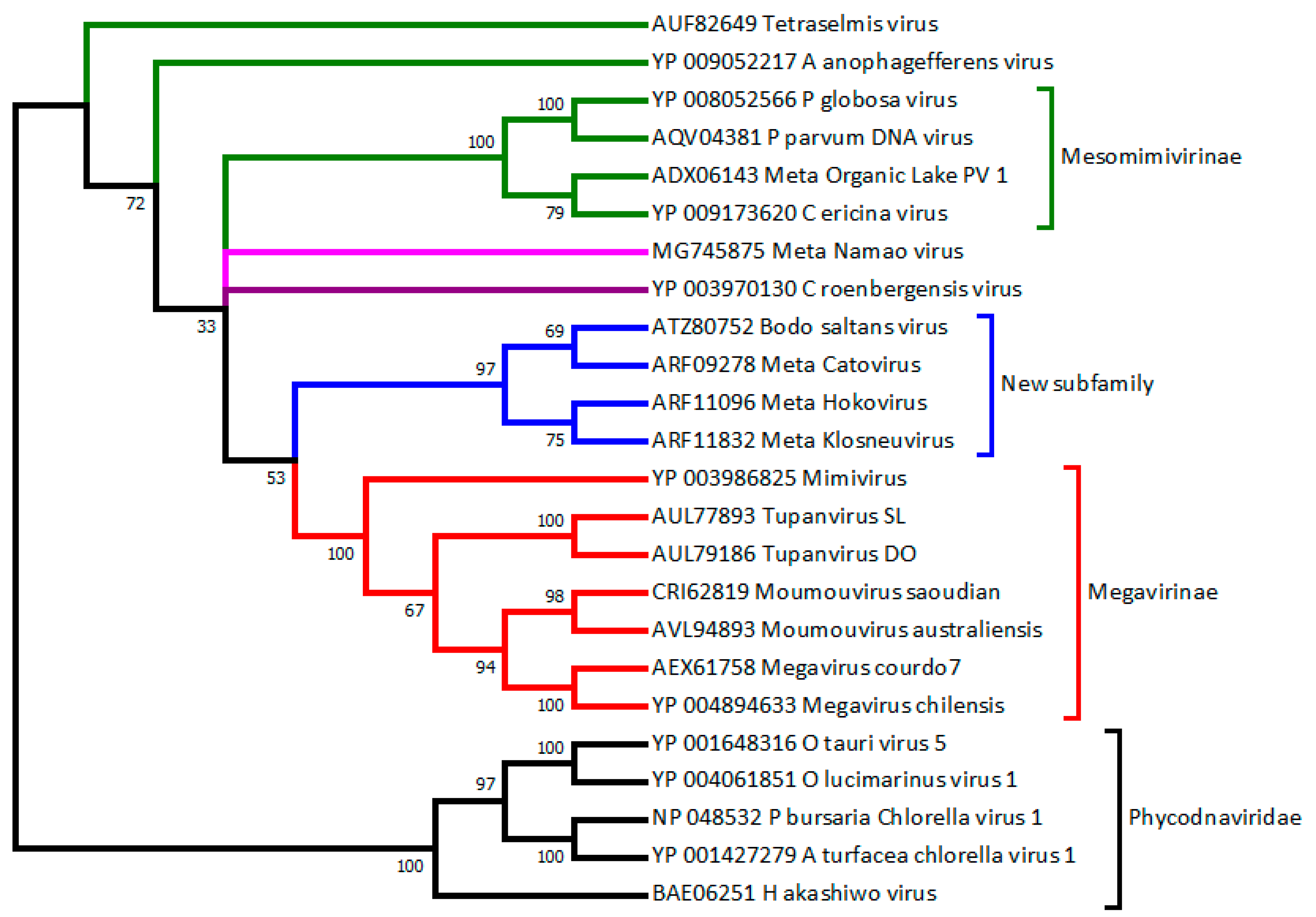
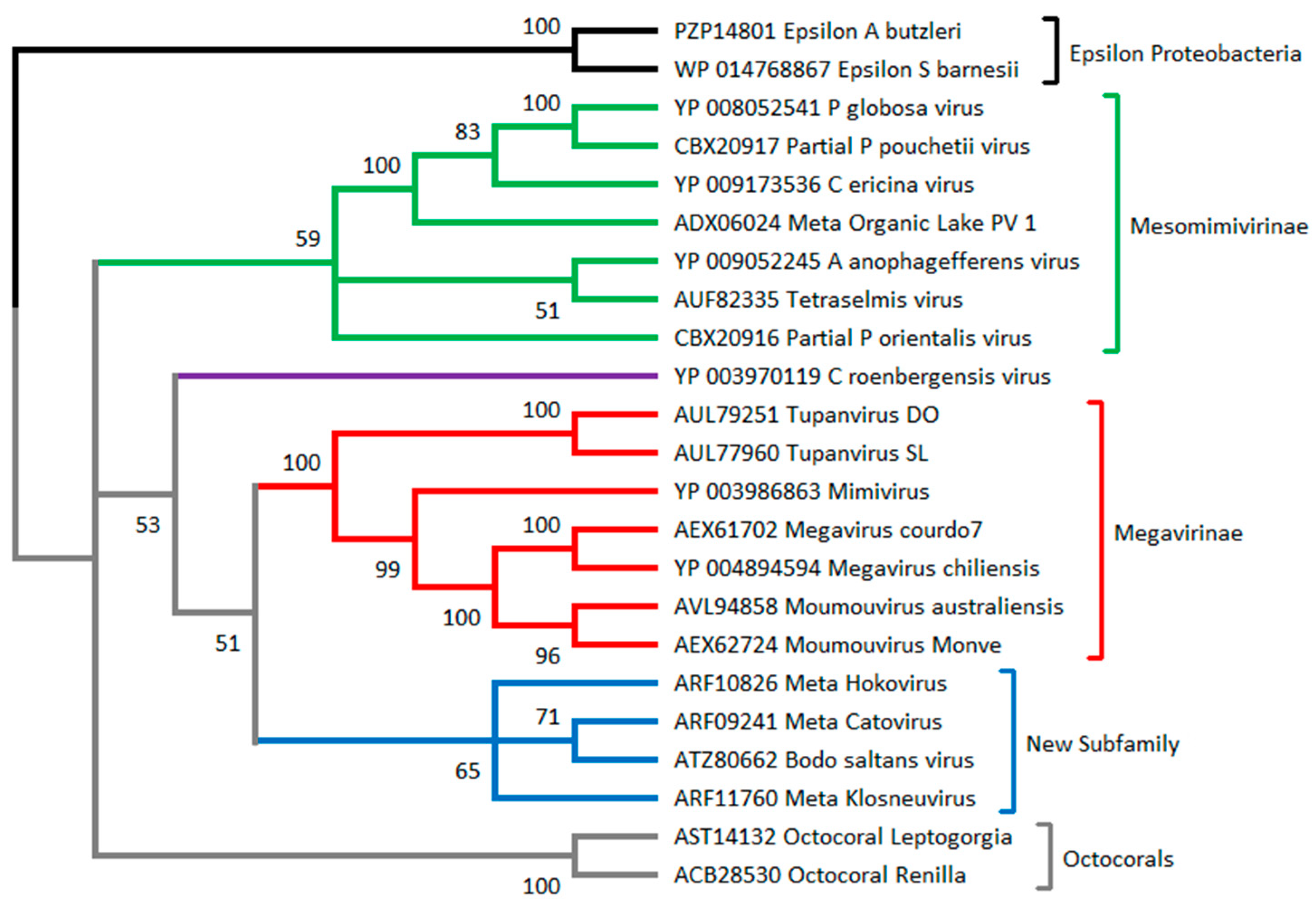
2.3. C. Roenbergensis Virus: The Prototype of a New Subfamily within the Mimiviridae
2.4. Smaller Mimiviridae Infecting Bona Fide Microalgae: Yet Another Subfamily
2.5. Recent Isolations of New Truly Giant Mimiviridae Members
2.6. Metagenomic Contributions to the Expansion of the Mimiviridae Family
3. Discussion
3.1. Algae-Infecting Mimiviridae versus Phycodnavirididae
3.2. How to Recognize and Classify Future Members of the Mimiviridae
- A high prevalence of the strictly conserved AAAATTGA motif in the promoter regions of early transcribed genes [85].
- A high prevalence of hairpin-forming transcription termination motifs in the 3′ end of genes [86].
- The detection of a transpoviron. Transpovirons are 7-kb-long plasmid-like linear dsDNA molecules found in association with Mimivirus-relatives [88].
- The presence of a gene encoding a Mimiviridae-specific version of glutamine-dependent Asparagine synthetase (AsnS). A different, easily distinguishable homolog of this enzyme is encoded by some Prasinoviruses (e.g., OtV5) [74].
- The presence of amino-acyl tRNA synthetases (aaRS). The finding of such central components of the translational apparatus in the Mimivirus genome [2] was considered revolutionary, as the historical definition of viruses denied them the capacity to synthetize proteins [4]. Conflicting hypotheses on the evolutionary origin of these viral enzymes generated a still ongoing controversial debate. The presence of these aaRS remains a remarkable feature of the Mimiviridae, although not unique to them anymore [7]. Unfortunately, the number of virus-encoded aaRS varies greatly among the Mimiviridae members (from 0 up to 20 different ones in Tupanviruses [46]) (Table 3), which reduces the utility of their detection for classification purpose. However, the presence of aaRS in a virus genome remains a strong complementary argument for their classification within the Mimiviridae.
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- La Scola, B.; Audic, S.; Robert, C.; Jungang, L.; de Lamballerie, X.; Drancourt, M.; Birtles, R.; Claverie, J.M.; Raoult, D. A giant virus in amoebae. Science 2003, 299, 2033. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; La Scola, B.; Suzan, M.; Claverie, J.M. The 1.2-megabase genome sequence of Mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; La Scola, B.; Birtles, R. The discovery and characterization of Mimivirus, the largest known virus and putative pneumonia agent. Clin. Infect. Dis. 2007, 45, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Abergel, C.; Legendre, M.; Claverie, J.M. The rapidly expanding universe of giant viruses: Mimivirus, Pandoravirus, Pithovirus and Mollivirus. FEMS Microbiol. Rev. 2015, 39, 779–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claverie, J.M.; Abergel, C. Giant viruses: The difficult breaking of multiple epistemological barriers. Stud. Hist. Philos. Biol. Biomed. Sci. 2016, 59, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Ogata, H.; Audic, S.; Abergel, C.; Suhre, K.; Fournier, P.E. Mimivirus and the emerging concept of “giant” virus. Virus Res. 2006, 117, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philippe, N.; Legendre, M.; Doutre, G.; Couté, Y.; Poirot, O.; Lescot, M.; Arslan, D.; Seltzer, V.; Bertaux, L.; Bruley, C.; et al. Pandoraviruses: Amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science 2013, 341, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, C.; Eisen, J.A.; Nene, V. New evolutionary frontiers from unusual virus genomes. Genome Biol. 2005, 6, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzan-Monti, M.; La Scola, B.; Raoult, D. Genomic and evolutionary aspects of Mimivirus. Virus Res. 2006, 117, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M. Viruses take center stage in cellular evolution. Genome Biol. 2006, 7, 110. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Forterre, P. Redefining viruses: Lessons from Mimivirus. Nat. Rev. Microbiol. 2008, 6, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Grzela, R.; Lartigue, A.; Bernadac, A.; Nitsche, S.; Vacelet, J.; Ogata, H.; Abergel, C. Mimivirus and Mimiviridae: Giant viruses with an increasing number of potential hosts, including corals and sponges. J. Invertebr. Pathol. 2009, 101, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Forterre, P. Giant viruses: Conflicts in revisiting the virus concept. Intervirology 2010, 53, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Abergel, C. Mimivirus: The emerging paradox of quasi-autonomous viruses. Trends Genet. 2010, 26, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Abergel, C. Open questions about giant viruses. Adv. Virus Res. 2013, 85, 25–56. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.; Brochier-Armanet, C. Giant viruses, giant chimeras: The multiple evolutionary histories of Mimivirus genes. BMC Evol. Biol. 2008, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.; Lopez-Garcia, P. Ten reasons to exclude viruses from the tree of life. Nat. Rev. Microbiol. 2009, 7, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Ludmir, E.B.; Enquist, L.W. Viral genomes are part of the phylogenetic tree of life. Nat. Rev. Microbiol. 2009, 7, 615. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Ogata, H. Ten good reasons not to exclude giruses from the evolutionary picture. Nat. Rev. Microbiol. 2009, 7, 615. [Google Scholar] [CrossRef] [PubMed]
- Hegde, N.R.; Maddur, M.S.; Kaveri, S.V.; Bayry, J. Reasons to include viruses in the tree of life. Nat. Rev. Microbiol. 2009, 7, 615. [Google Scholar] [CrossRef] [PubMed]
- Navas-Castillo, J. Six comments on the ten reasons for the demotion of viruses. Nat. Rev. Microbiol. 2009, 7, 615. [Google Scholar] [CrossRef] [PubMed]
- López-García, P.; Moreira, D. Yet viruses cannot be included in the tree of life. Nat. Rev. Microbiol. 2009, 7, 615–617. [Google Scholar] [CrossRef]
- Boyer, M.; Madoui, M.A.; Gimenez, G.; La Scola, B.; Raoult, D. Phylogenetic and phyletic studies of informational genes in genomes highlight existence of a 4 domain of life including giant viruses. PLoS ONE 2010, 5, e15530. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Arslan, D.; Abergel, C.; Claverie, J.M. Genomics of Megavirus and the elusive fourth domain of Life. Commun. Integr. Biol. 2012, 5, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Nasir, A.; Kim, K.M.; Caetano-Anolles, G. Giant viruses coexisted with the cellular ancestors and represent a distinct supergroup along with superkingdoms Archaea, Bacteria and Eukarya. BMC Evol. Biol. 2012, 12, 156. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Senkevich, T.G.; Dolja, V.V. Compelling reasons why viruses are relevant for the origin of cells. Nat. Rev. Microbiol. 2009, 7, 615. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, L.P.; Witzany, G. Viruses are essential agents within the roots and stem of the tree of life. J. Theor. Biol. 2010, 262, 698–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasir, A.; Sun, F.J.; Kim, K.M.; Caetano-Anollés, G. Untangling the origin of viruses and their impact on cellular evolution. Ann. N. Y. Acad. Sci. 2015, 1341, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Beijerinck, M.W. Über ein Contagium vivum fluidum als Ursache der Fleckenkrankheit der Tabaksblätter. Verhandelingen der Koninklijke akademie van Wetenschappen te Amsterdam 1898, 65, 1–22. [Google Scholar]
- Lecoq, H. Discovery of the first virus, the tobacco mosaic virus: 1892 or 1898? C. R. Acad. Sci. III 2001, 324, 929–933. [Google Scholar] [CrossRef]
- Meints, R.H.; Van Etten, J.L.; Kuczmarski, D.; Lee, K.; Ang, B. Viral infection of the symbiotic chlorella-like alga present in Hydra viridis. Virology 1981, 113, 698–703. [Google Scholar] [CrossRef]
- Van Etten, J.L.; Meints, R.H.; Burbank, D.E.; Kuczmarski, D.; Cuppels, D.A.; Lane, L.C. Isolation and characterization of a virus from the intracellular green alga symbiotic with Hydra viridis. Virology 1981, 113, 704–771. [Google Scholar] [CrossRef]
- Torrella, F.; Morita, R.Y. Evidence by electron micrographs for a high incidence of bacteriophage particles in the waters of Yaquina Bay, oregon: Ecological and taxonomical implications. Appl. Environ. Microbiol. 1979, 37, 774–778. [Google Scholar] [PubMed]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, Z.; Sun, L.; Ropp, S.; Kutish, G.F.; Rock, D.L.; Van Etten, J.L. Analysis of 74 kb of DNA located at the right end of the 330-kb chlorella virus PBCV-1 genome. Virology 1997, 237, 360–377. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.L.; Meints, R.H.; Kuczmarski, D.; Burbank, D.E.; Lee, K. Viruses of symbiotic Chlorella-like algae isolated from Paramecium bursaria and Hydra viridis. Proc. Natl. Acad. Sci. USA 1982, 79, 3867–3871. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.L. Giant Chlorella viruses. Mol. Cells (Korean Soc. Mol. Cell. Biol.) 1995, 5, 99–106. [Google Scholar]
- Van Etten, J.L.; Meints, R.H. Giant viruses infecting algae. Annu. Rev. Microbiol. 1999, 53, 447–494. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M. Giant viruses in the oceans: The 4th Algal Virus Workshop. Virol. J. 2005, 2, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghedin, E.; Claverie, J.M. Mimivirus relatives in the Sargasso Sea. Virol. J. 2005, 2, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monier, A.; Claverie, J.M.; Ogata, H. Taxonomic distribution of large DNA viruses in the sea. Genome Biol. 2008, 9, R106. [Google Scholar] [CrossRef] [PubMed]
- Monier, A.; Larsen, J.B.; Sandaa, R.A.; Bratbak, G.; Claverie, J.M.; Ogata, H. Marine mimivirus relatives are probably large algal viruses. Virol. J. 2008, 5, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, H.; Ray, J.; Toyoda, K.; Sandaa, R.A.; Nagasaki, K.; Bratbak, G.; Claverie, J.M. Two new subfamilies of DNA mismatch repair proteins (MutS) specifically abundant in the marine environment. ISME J. 2011, 5, 1143–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arslan, D.; Legendre, M.; Seltzer, V.; Abergel, C.; Claverie, J.M. Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proc. Natl. Acad. Sci. USA 2011, 108, 17486–17491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrahão, J.; Silva, L.; Silva, L.S.; Khalil, J.Y.B.; Rodrigues, R.; Arantes, T.; Assis, F.; Boratto, P.; Andrade, M.; Kroon, E.G.; et al. Tailed giant Tupanvirus possesses the most complete translational apparatus of the known virosphere. Nat. Commun. 2018, 9, 749. [Google Scholar] [CrossRef] [PubMed]
- Garza, D.R.; Suttle, C.A. Large double-stranded DNA viruses which cause the lysis of a marine heterotrophic nanoflagellate (Bodo sp.) occur in natural marine viral communities. Aquat. Microb. Ecol. 1995, 9, 203–210. [Google Scholar] [CrossRef]
- Fischer, M.G.; Allen, M.J.; Wilson, W.H.; Suttle, C.A. Giant virus with a remarkable complement of genes infects marine zooplankton. Proc. Natl. Acad. Sci. USA 2010, 107, 19508–19513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, T.A.; Cavalier-Smith, T. Myosin domain evolution and the primary divergence of eukaryotes. Nature 2005, 436, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legendre, M.; Fabre, E.; Poirot, O.; Jeudy, S.; Lartigue, A.; Alempic, J.M.; Beucher, L.; Philippe, N.; Bertaux, L.; Christo-Foroux, E.; et al. Diversity and evolution of the emerging Pandoraviridae family. Nat. Commun. 2018, 9, 2285. [Google Scholar] [CrossRef] [PubMed]
- Sandaa, R.A.; Heldal, M.; Castberg, T.; Thyrhaug, R.; Bratbak, G. Isolation and characterization of two viruses with large genome size infecting Chrysochromulina ericina (Prymnesiophyceae) and Pyramimonas orientalis (Prasinophyceae). Virology 2001, 290, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Brussaard, C.P.; Short, S.M.; Frederickson, C.M.; Suttle, C.A. Isolation and phylogenetic analysis of novel viruses infecting the phytoplankton Phaeocystis globose (Prymnesiophyceae). Appl. Environ. Microbiol. 2004, 70, 3700–3705. [Google Scholar] [CrossRef] [PubMed]
- Baudoux, A.C.; Brussaard, C.P. Characterization of different viruses infecting the marine harmful algal bloom species Phaeocystis globosa. Virology 2005, 341, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.B.; Larsen, A.; Bratbak, G.; Sandaa, R.A. Phylogenetic analysis of members of the Phycodnaviridae virus family, using amplified fragments of the major capsid protein gene. Appl. Environ. Microbiol. 2008, 74, 3048–3057. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.; Jeudy, S.; Bartoli, J.; Poirot, O.; Lescot, M.; Abergel, C.; Barbe, V.; Wommack, K.E.; Noordeloos, A.A.; Brussaard, C.P.; et al. Genome of Phaeocystis globosa virus PgV-16T highlights the common ancestry of the largest known DNA viruses infecting eukaryotes. Proc. Natl. Acad. Sci. USA 2013, 110, 10800–10805. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; LeCleir, G.R.; Brown, C.M.; Gobler, C.J.; Bidle, K.D.; Wilson, W.H.; Wilhelm, S.W. Genome of brown tide virus (AaV), the little giant of the Megaviridae, elucidates NCLDV genome expansion and host-virus coevolution. Virology 2014, 466–467, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Gallot-Lavallée, L.; Blanc, G.; Claverie, J.M. Comparative Genomics of Chrysochromulina Ericina Virus and Other Microalga-Infecting Large DNA Viruses Highlights Their Intricate Evolutionary Relationship with the Established Mimiviridae Family. J. Virol. 2017, 91, JVI-00230. [Google Scholar] [CrossRef] [PubMed]
- Schvarcz, C.R.; Steward, G.F. A giant virus infecting green algae encodes key fermentation genes. Virology 2018, 518, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Sicko-Goad, L.; Walker, G. Viroplasm and large virus-like particles in the dinoflagellate Gymnodinium uberrimum. Protoplasma 1979, 99, 203–210. [Google Scholar] [CrossRef]
- Dodds, J.A.; Cole, A. Microscopy and biology of Uronema gigas, a filamentous eucaryotic green alga, and its associated tailed virus-like particle. Virology 1980, 100, 156–165. [Google Scholar] [CrossRef]
- Deeg, C.M.; Chow, C.T.; Suttle, C.A. The kinetoplastid-infecting Bodo saltans virus (BsV), a window into the most abundant giant viruses in the sea. eLife 2018, 7, e33014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, S.J.; Rusch, D.B.; Yooseph, S.; Halpern, A.L.; Heidelberg, K.B.; Glass, J.I.; Andrews-Pfannkoch, C.; Fadrosh, D.; Miller, C.S.; Sutton, G.; et al. The Sorcerer II Global Ocean Sampling Expedition: Metagenomic characterization of viruses within aquatic microbial samples. PLoS ONE 2008, 3, e1456. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Abergel, C.; Ogata, H. Mimivirus. Curr. Top Microbiol. Immunol. 2009, 328, 89–121. [Google Scholar] [PubMed]
- Yau, S.; Lauro, F.M.; DeMaere, M.Z.; Brown, M.V.; Thomas, T.; Raftery, M.J.; Andrews-Pfannkoch, C.; Lewis, M.; Hoffman, J.M.; Gibson, J.A.; et al. Virophage control of antarctic algal host-virus dynamics. Proc. Natl. Acad. Sci. USA 2011, 108, 6163–6168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Scola, B.; Desnues, C.; Pagnier, I.; Robert, C.; Barrassi, L.; Fournous, G.; Merchat, M.; Suzan-Monti, M.; Forterre, P.; Koonin, E.; et al. The virophage as a unique parasite of the giant mimivirus. Nature 2008, 455, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Abergel, C. Mimivirus and its virophage. Annu. Rev. Genet. 2009, 43, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, W.; Yan, S.; Xiao, J.; Zhang, Y.; Li, B.; Pan, Y.; Wang, Y. Diversity of virophages in metagenomic data sets. J. Virol. 2013, 87, 4225–4236. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Sun, D.; Childers, A.; McDermott, T.R.; Wang, Y.; Liles, M.R. Three novel virophage genomes discovered from Yellowstone Lake metagenomes. J. Virol. 2015, 89, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Bekliz, M.; Colson, P.; La Scola, B. The Expanding Family of Virophages. Viruses 2016, 8, 317. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Chan, L.K.; Egan, R.; Malmstrom, R.R.; McMahon, K.D.; Sullivan, M.B. Ecogenomics of virophages and their giant virus hosts assessed through time series metagenomics. Nat. Commun. 2017, 8, 858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsenti, E.; Acinas, S.G.; Bork, P.; Bowler, C.; de Vargas, C.; Raes, J.; Sullivan, M.; Arendt, D.; Benzoni, F.; Claverie, J.M.; et al. A holistic approach to marine eco-systems biology. PLoS Biol. 2011, 9, e1001177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingamp, P.; Grimsley, N.; Acinas, S.G.; Clerissi, C.; Subirana, L.; Poulain, J.; Ferrera, I.; Sarmento, H.; Villar, E.; Lima-Mendez, G.; et al. Exploring nucleo-cytoplasmic large DNA viruses in Tara Oceans microbial metagenomes. ISME J. 2013, 7, 1678–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mozar, M.; Claverie, J.M. Expanding the Mimiviridae family using asparagine synthase as a sequence bait. Virology 2014, 466–467, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Schulz, F.; Yutin, N.; Ivanova, N.N.; Ortega, D.R.; Lee, T.K.; Vierheilig, J.; Daims, H.; Horn, M.; Wagner, M.; Jensen, G.J.; et al. Giant viruses with an expanded complement of translation system components. Science 2017, 356, 82–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clouthier, S.C.; Vanwalleghem, E.; Copeland, S.; Klassen, C.; Hobbs, G.; Nielsen, O.; Anderson, E.D. A new species of nucleo-cytoplasmic large DNA virus (NCLDV) associated with mortalities in Manitoba lake sturgeon Acipenser fulvescens. Dis. Aquat. Organ. 2013, 102, 195–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clouthier, S.C.; Anderson, E.D.; Kurath, G.; Breyta, R. Molecular systematics of sturgeon nucleocytoplasmic large DNA viruses. Mol. Phylogenet. Evol. 2018, 128, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Maumus, F.; Epert, A.; Nogué, F.; Blanc, G. Plant genomes enclose footprints of past infections by giant virus relatives. Nat. Commun. 2014, 5, 4268. [Google Scholar] [CrossRef] [PubMed]
- Maumus, F.; Blanc, G. Study of Gene Trafficking between Acanthamoeba and Giant Viruses Suggests an Undiscovered Family of Amoeba-Infecting Viruses. Genome Biol. Evol. 2016, 8, 3351–3363. [Google Scholar] [CrossRef] [PubMed]
- Brister, J.R.; Ako-Adjei, D.; Bao, Y.; Blinkova, O. NCBI viral genomes resource. Nucleic Acids Res. 2015, 43, D571–D577. [Google Scholar] [CrossRef] [PubMed]
- Pagarete, A.; Grébert, T.; Stepanova, O.; Sandaa, R.A.; Bratbak, G. Tsv-N1: A Novel DNA Algal Virus that Infects Tetraselmis striata. Viruses 2015, 7, 3937–3953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, H.; Toyoda, K.; Tomaru, Y.; Nakayama, N.; Shirai, Y.; Claverie, J.M.; Nagasaki, K. Remarkable sequence similarity between the dinoflagellate-infecting marine girus and the terrestrial pathogen African swine fever virus. Virol. J. 2009, 6, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackinder, L.C.; Worthy, C.A.; Biggi, G.; Hall, M.; Ryan, K.P.; Varsani, A.; Harper, G.M.; Wilson, W.H.; Brownlee, C.; Schroeder, D.C. A unicellular algal virus, Emiliania huxleyi virus 86, exploits an animal-like infection strategy. J. Gen. Virol. 2009, 90, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Colson, P.; Raoult, D.; Koonin, E.V. Mimiviridae: Clusters of orthologous genes, reconstruction of gene repertoire evolution and proposed expansion of the giant virus family. Virol. J. 2013, 10, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suhre, K.; Audic, S.; Claverie, J.M. Mimivirus gene promoters exhibit an unprecedented conservation among all eukaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 14689–14693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, D.; Grzela, R.; Lartigue, A.; Audic, S.; Chenivesse, S.; Encinas, S.; Claverie, J.M.; Abergel, C. The polyadenylation site of Mimivirus transcripts obeys a stringent ‘hairpin rule’. Genome Res. 2009, 19, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.G.; Suttle, C.A. A virophage at the origin of large DNA transposons. Science 2011, 332, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Desnues, C.; La Scola, B.; Yutin, N.; Fournous, G.; Robert, C.; Azza, S.; Jardot, P.; Monteil, S.; Campocasso, A.; Koonin, E.V.; et al. Provirophages and transpovirons as the diverse mobilome of giant viruses. Proc. Natl. Acad. Sci. USA 2012, 109, 18078–18083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihara, T.; Koyano, H.; Hingamp, P.; Grimsley, N.; Goto, S.; Ogata, H. Taxon Richness of “Megaviridae” Exceeds those of Bacteria and Archaea in the Ocean. Microbes Environ. 2018, 33, 162–171. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Name 1 | Accession | Genome Size (kb) | Virion Type | Virion Size (nm) | Host Phylum |
|---|---|---|---|---|---|
| Mimivirus 2 | NC_014649 | 1181 | icosahedron | 750 | Amoebozoa |
| Megavirus 2 | NC_016072 | 1259 | icosahedron | 680 | Amoebozoa |
| Moumouvirus 2 | NC_020104 | 1021 | icosahedron | 620 | Amoebozoa |
| CroV | NC_014637 | 693 | icosahedron | 300 | Heterokonta |
| PgV | NC_021312 | 460 | icosahedron | 150 | Haptophyceae |
| CeV | NC_028094 | 474 | icosahedron | 160 | Haptophyceae |
| TetV | KY322437 | 668 | icosahedron | 240 | Chlorophyta |
| BsV | MF782455 | 1386 | icosahedron | 300 | Excavata |
| AaV | NC_024697 | 371 | icosahedron | 140 | Heterokonta |
| TupanSL | KY523104 | 1439 | icosa. + tail | 450 + 550 | Amoebozoa |
| TupanDO | MF405918 | 1516 | icosa. + tail | 450 + 550 | Amoebozoa |
| Genus | Prototype | Encoded RNA pol | Genome Size (kb) | (G + C) % | Accession | Virion Ø (nm) |
|---|---|---|---|---|---|---|
| Chlorovirus | PBCV-1 | No | 330 | 40 | NC_000852 | 190 |
| Coccolithovirus 1 | EhV-86 | Yes | 407 | 40.2 | NC_007346 | 180 |
| Phaeovirus | EsV-1 | No | 336 | 51.7 | NC_002687 | 200 |
| Prasinovirus | OtV-5 | No | 187 | 45 | NC_010191 | 120 |
| Prymnesiovirus I 2 | PgV-16T | Yes | 460 | 32 | NC_021312 | 153 |
| Prymnesiovirus II 2 | PgV-01T | Unknown | ≈177 | Unknown | - | 106 |
| Raphidovirus | HaV-1 | No | 275 | 30.4 | KX008963 | 202 |
| Name | DNA Pol B | RNA 2 Pol II | aaRS 3 | MCP 4 | MutS7 | AsnS | Transpoviron | Virophage |
|---|---|---|---|---|---|---|---|---|
| Mimivirus | Yes | 8 | 4 | 4 | Yes | Yes | Yes | Yes |
| Megavirus | Yes | 8 | 7 | 4 | Yes | Yes | Yes | Yes |
| Moumouvirus | Yes | 8 | 5 | 4 | Yes | Yes | Yes | Yes |
| CroV | Yes | 8 | 1 | 4 | Yes | Yes | No | Yes |
| PgV | Yes | 8 | 0 | 2 | Yes | Yes | No | Yes |
| CeV | Yes | 8 | 0 | 3 | Yes | Yes | No | No |
| TetV | Yes | 8 | 0 | 1 | Yes | No | No | No |
| BsV | Yes | 7 | 2 | 4 | Yes | Yes | No | No |
| AaV | Yes | 8 | 0 | 2 | Yes | No | No | No |
| TupanSL | Yes | 8 | 20 | 3 | Yes | Yes | No | No |
| TupanDO | Yes | 7 | 20 | 3 | Yes | Yes | No | No |
| YlmV 1 | No | D | 0 | 1 | No | No | No | No |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claverie, J.-M.; Abergel, C. Mimiviridae: An Expanding Family of Highly Diverse Large dsDNA Viruses Infecting a Wide Phylogenetic Range of Aquatic Eukaryotes. Viruses 2018, 10, 506. https://doi.org/10.3390/v10090506
Claverie J-M, Abergel C. Mimiviridae: An Expanding Family of Highly Diverse Large dsDNA Viruses Infecting a Wide Phylogenetic Range of Aquatic Eukaryotes. Viruses. 2018; 10(9):506. https://doi.org/10.3390/v10090506
Chicago/Turabian StyleClaverie, Jean-Michel, and Chantal Abergel. 2018. "Mimiviridae: An Expanding Family of Highly Diverse Large dsDNA Viruses Infecting a Wide Phylogenetic Range of Aquatic Eukaryotes" Viruses 10, no. 9: 506. https://doi.org/10.3390/v10090506
APA StyleClaverie, J.-M., & Abergel, C. (2018). Mimiviridae: An Expanding Family of Highly Diverse Large dsDNA Viruses Infecting a Wide Phylogenetic Range of Aquatic Eukaryotes. Viruses, 10(9), 506. https://doi.org/10.3390/v10090506