Double-Stranded RNA High-Throughput Sequencing Reveals a New Cytorhabdovirus in a Bean Golden Mosaic Virus-Resistant Common Bean Transgenic Line

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. dsRNA Extraction
2.3. High-Throughput Sequencing and Assembly
2.4. Viral-Specific RT-PCR, Cloning, and Sequencing
2.5. 5′/3′ Rapid Amplification of cDNA Ends (RACE)
2.6. Phylogenetic Analysis and Pairwise Comparison
3. Results and Discussion
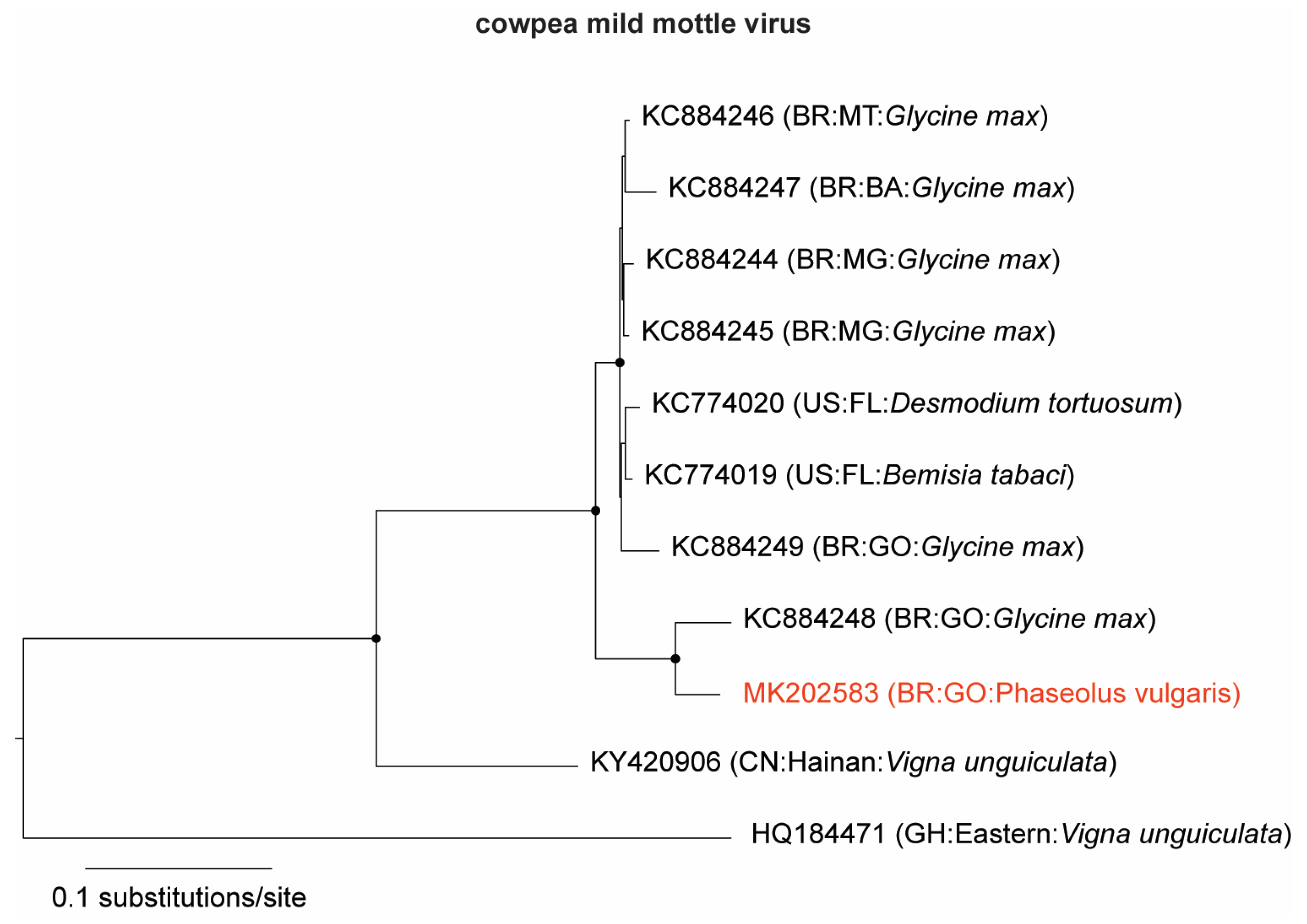
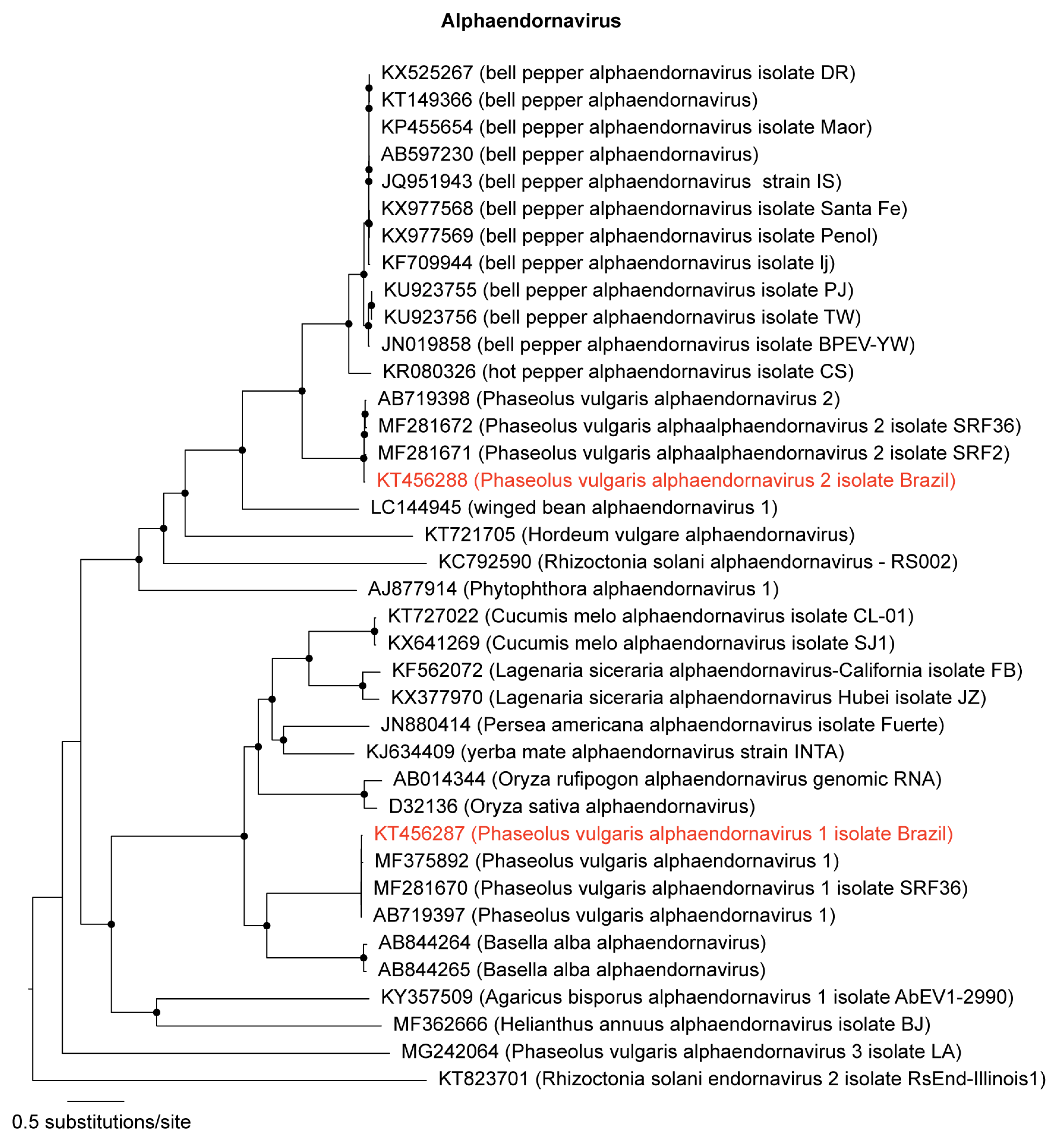
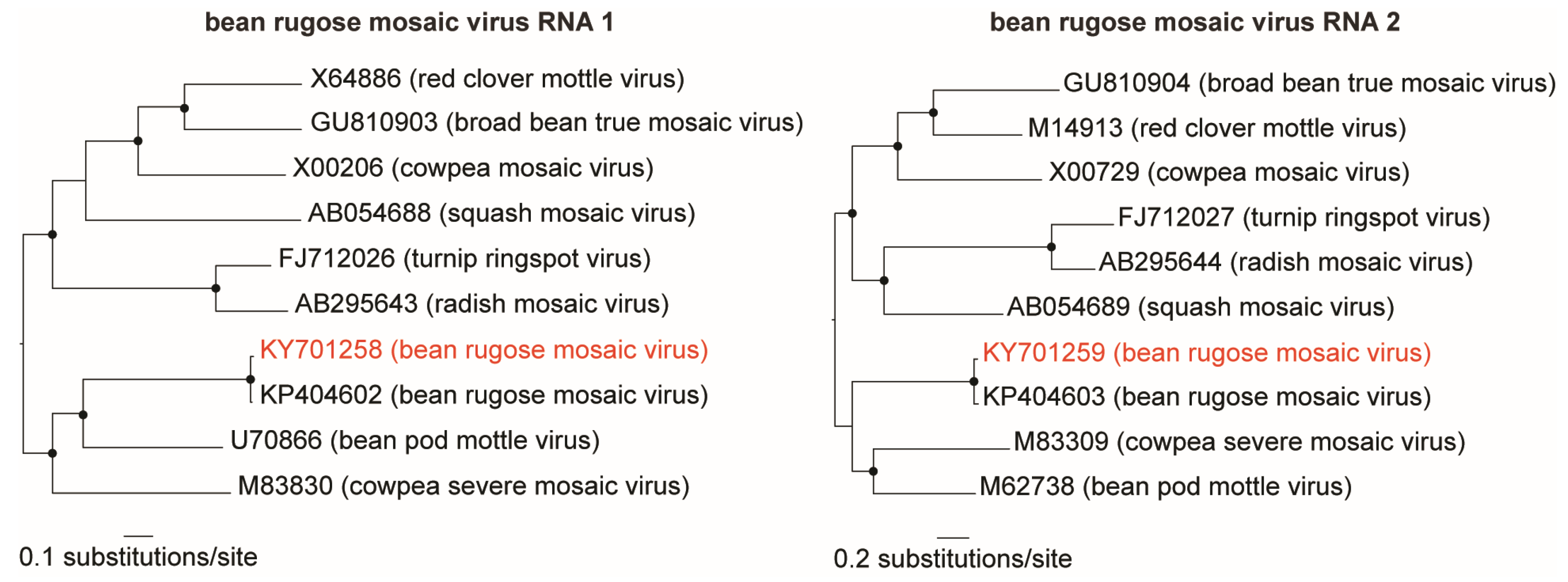
3.1. Identification and Genome Assembly of Known Bean-Infecting Viruses
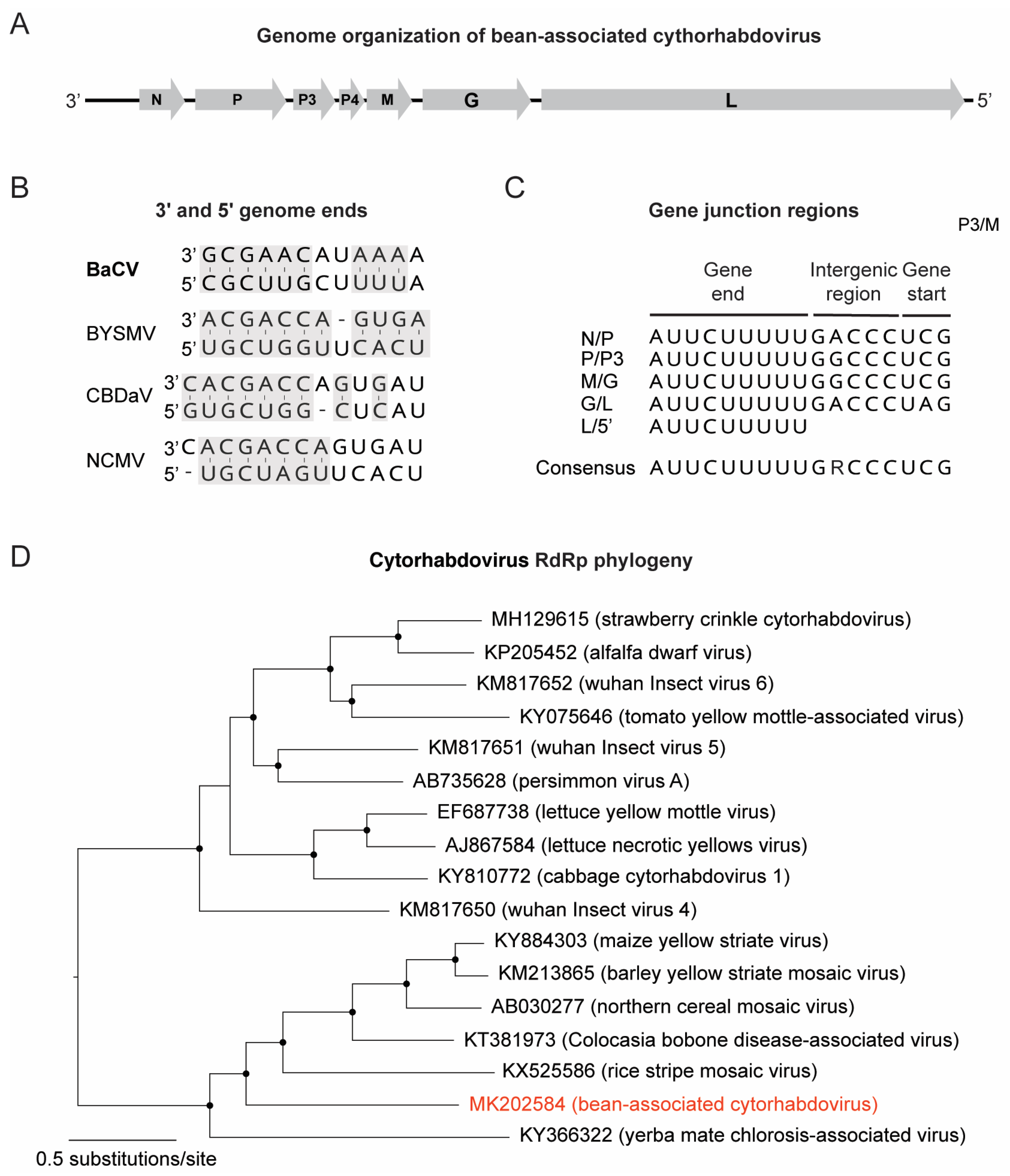
3.2. Discovery of a Novel Cytorhabdovirus
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- FAOSTAT. Food and Agriculture Organization of the United Nations-Statistics Division. 2018. Available online: http://www.fao.org/faostat/en/#home (accessed on 14 December 2018).
- Faria, J.C.; Aragão, F.J.L.; Souza, T.L.P.O.; Quintela, E.D.; Kitajima, E.W.; Ribeiro, S.G. Golden Mosaic of Common Beans in Brazil: Management with a Transgenic Approach. APS Features 2016. [Google Scholar] [CrossRef]
- Okada, R.; Yong, C.K.; Valverde, R.A.; Sabanadzovic, S.; Aoki, N.; Hotate, S.; Kiyota, E.; Moriyama, H.; Fukuhara, T. Molecular characterization of two evolutionarily distinct endornaviruses co-infecting common bean (Phaseolus vulgaris). J. Gen. Virol. 2013, 94, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, R.L.; Faria, J.C.; Ahlquist, P.; Maxwell, D.P. Genetic Diversity in Geminiviruses Causing Bean Golden Mosaic Disease: The Nucleotide-Sequence of the Infectious Cloned DNA Components of a Brazilian Isolate of Bean Golden Mosaic Geminivirus. Phytopathology 1993, 83, 709–715. [Google Scholar] [CrossRef]
- Faria, J.C.; Gilbertson, R.L.; Hanson, S.F.; Morales, F.J.; Ahlquist, P.; Loniello, A.O.; Maxwell, D.P. Bean golden mosaic geminivirus type II isolates from the Dominican Republic and Guatemala: Nucleotide sequences, infectious pseudorecombinants and phylogenetic relationships. Phytopathology 1994, 84, 321–329. [Google Scholar] [CrossRef]
- Worrall, E.A.; Wamonje, F.O.; Mukeshimana, G.; Harvey, J.J.W.; Carr, J.P.; Mitter, N. Chapter One—Bean Common Mosaic Virus and Bean Common Mosaic Necrosis Virus: Relationships, Biology, and Prospects for Control. In Advances in Virus Research; Kielian, M., Maramorosch, K., Mettenleiter, T.C., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 93, pp. 1–46. [Google Scholar]
- Marubayashi, J.M.; Yuki, V.A.; Wutke, E.B. Transmissão do Cowpea mild mottle virus pela mosca branca Bemisia tabaci biótipo B para plantas de feijão e soja. Summa Phytopathol. 2010, 36, 158–160. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.S. Three whitefly-transmitted virus diseases of beans in São Paulo, Brazil. Plant Prot. Bull. FAO 1965, 13, 2–12. [Google Scholar]
- Bonfim, K.; Faria, J.C.; Nogueira, E.O.P.L.; Mendes, E.A.; Aragao, F.J.L. RNAi-Mediated Resistance to Bean golden mosaic virus in Genetically Engineered Common Bean (Phaseolus vulgaris). Mol. Plant Microbe Interact. 2007, 20, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Aragao, F.J.; Faria, J.C. First transgenic geminivirus-resistant plant in the field. Nat. Biotechnol. 2009, 27, 1086–1088. [Google Scholar] [CrossRef] [PubMed]
- Mascia, T.; Gallitelli, D. Synergies and antagonisms in virus interactions. Plant Sci. 2016, 252, 176–192. [Google Scholar] [CrossRef]
- Syller, J. Facilitative and antagonistic interactions between plant viruses in mixed infections. Mol. Plant Pathol. 2012, 13, 204–216. [Google Scholar] [CrossRef]
- Melcher, U.; Ali, A. Virus–Virus Interactions in Plants. In Plant Viruses: Diversity, Interaction and Management; Gaur, R.K., SMP, K., Dorokhov, Y., Eds.; Taylor & Francis Group: New York, NY, USA, 2018; pp. 249–266. [Google Scholar]
- Valverde, R.A.; Nameth, S.T.; Jordon, R.L. Analysis of Double-Stranded RNA for Plant Virus Diagnosis. Plant Dis. 1990, 74, 255–258. [Google Scholar]
- Castillo, A.; Cottet, L.; Castro, M.; Sepulveda, F. Rapid isolation of mycoviral double-stranded RNA from Botrytis cinerea and Saccharomyces cerevisiae. Virol. J. 2011, 8, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Frohman, M.A.; Dush, M.K.; Martin, G.R. Rapid production of full-length cDNAs from rare transcripts: Amplification using a single gene-specific oligonucleotide primer. Proc. Nati. Acad. Sci. USA 1988, 85, 8998–9002. [Google Scholar] [CrossRef]
- Nicolini, C.; Pio-Ribeiro, G.; Andrade, G.P.; Melo, F.L.; Oliveira, V.C.; Guimaraes, F.C.; Resende, R.O.; Kitajima, E.W.; Rezende, J.A.; Nagata, T. A distinct tymovirus infecting Cassia hoffmannseggii in Brazil. Virus Genes 2012, 45, 190–194. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H.; Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 1999, 16, 1114–1116. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v1.4.4. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 26 September 2018).
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Nordenstedt, N.; Marcenaro, D.; Chilagane, D.; Mwaipopo, B.; Rajamaki, M.L.; Nchimbi-Msolla, S.; Njau, P.J.R.; Mbanzibwa, D.R.; Valkonen, J.P.T. Pathogenic seedborne viruses are rare but Phaseolus vulgaris endornaviruses are common in bean varieties grown in Nicaragua and Tanzania. PLoS ONE 2017, 12, e0178242. [Google Scholar]
- Wainaina, J.M.; Ateka, E.; Makori, T.; Kehoe, M.A.; Boykin, L.M. Phylogenetic relationships of endornaviruses in common bean from the western highlands of Kenya and global sequences. PeerJ Preprints 2018, 6, e26904v1. [Google Scholar]
- Zanardo, L.G.; Silva, F.N.; Lima, A.T.; Milanesi, D.F.; Castilho-Urquiza, G.P.; Almeida, A.M.; Zerbini, F.M.; Carvalho, C.M. Molecular variability of cowpea mild mottle virus infecting soybean in Brazil. Arch. Virol. 2014, 159, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Capobianco, H.; Ng, T.F.; Breitbart, M.; Polston, J.E. RNA viral metagenome of whiteflies leads to the discovery and characterization of a whitefly-transmitted carlavirus in North America. PLoS ONE 2014, 9, e86748. [Google Scholar] [CrossRef] [PubMed]
- Picoli, M.H.; Garcia, A.; Barboza, A.A.; de Souto, E.R.; Almeida, A.M. Complete genome sequence of bean rugose mosaic virus, genus Comovirus. Arch. Virol. 2016, 161, 1711–1714. [Google Scholar] [CrossRef]
- Walker, P.J.; Blasdell, K.R.; Calisher, C.H.; Dietzgen, R.G.; Kondo, H.; Kurath, G.; Longdon, B.; Stone, D.M.; Tesh, R.B.; Tordo, N.; et al. ICTV Virus Taxonomy Profile: Rhabdoviridae. J. Gen. Virol. 2018, 99, 447–448. [Google Scholar] [CrossRef] [Green Version]
- Bejerman, N.; de Breuil, S.; Debat, H.; Miretti, M.; Badaracco, A.; Nome, C. Molecular characterization of yerba mate chlorosis-associated virus, a putative cytorhabdovirus infecting yerba mate (Ilex paraguariensis). Arch. Virol. 2017, 162, 2481–2484. [Google Scholar] [CrossRef]
- Walker, P.J.; Firth, C.; Widen, S.G.; Blasdell, K.R.; Guzman, H.; Wood, T.G.; Paradkar, P.N.; Holmes, E.C.; Tesh, R.B.; Vasilakis, N. Evolution of genome size and complexity in the rhabdoviridae. PLoS Pathog. 2015, 11, e1004664. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Primer Name | Sequence 5′-3′ | Amplicon (bp) | Tm (°C) |
|---|---|---|---|---|
| Alphaendornavirus | PvEV-1 1 F | GTAAACCAGGGAATTGGTGG 5 | 303 | 60 |
| PvEV-1 1 R | GATTGATTGGGCTGTATAGTG 5 | |||
| PvEV-2 1 F | TGTTAGGCGTGTGTCCCCA 5 | 519 | 56 | |
| PvEv-2 1 R | GTTGCTGTATTGCTCGTGTC 5 | |||
| Comovirus | BRMV 2-RNA1-2550 F | GACAATACAGCCTATGATGGGA | 995 | 61 |
| BRMV 2-RNA1-3545 R | CACCAATGATCCACAATCCA | |||
| BRMV 2-RNA2-1704 F | TCTGGTGATGGGTTATTTTCTCAGA | 1832 | 60 | |
| BRMV 2-RNA2-3536 R | CTACTGATACATCCTATCCATTGCA | |||
| Carlavirus | CPMMV 3-4000 F | AACTTGGCCTTAGTGAACTCTACA | 500 | 58 |
| CPMMV 3-4500 R | ATTAGCTCTGTGCCTGGGGT | |||
| Cytorhabdovirus | BaCV 4-6491 F | GAAGTCGCATAGCTCGTCGA | 687 | 60 |
| BaCV 4-7178 R | GAGCGATAAGAACCTCCCCG |
| GenBank Accession Number | Length (nt) | Mapped Reads | % of Total Reads | Coverage | Classification | ||
|---|---|---|---|---|---|---|---|
| Genus | Virus | Isolate | |||||
| MK202583 | 8194 | 1,131,964 | 8.2 | 19,810× | Carlavirus | CPMMV 1 | CPMMV:BR:GO:14 |
| KT456287 | 14,072 | 73,996 | 0.5 | 762× | Alphaendornavirus | PvEV-1 2 | PvEV-1 (Brazil) |
| KT456288 | 14,817 | 114,256 | 0.8 | 1123× | Alphaendornavirus | PvEV-2 2 | PvEV-2 (Brazil) |
| KY701258 | 5891 | 151,744 | 1.1 | 3500× | Comovirus | BRMV-RNA1 3 | BRMV- BR-GO |
| KY701259 | 3735 | 182,331 | 1.3 | 7000× | BRMV-RNA2 3 | BRMV- BR-GO | |
| MK202584 | 13,467 | 2400 | 0.01 | 26× | Cytorhabdovirus | BaCV 4 | BacV-BR-GO |
| ORFs 1 | Size (nt/aa) | Putative Product | Blastp Hit (Organism) (Acession) |
|---|---|---|---|
| 1 | 1356/451 | Nucleoprotein (N) | Nucleocapsid protein (RSMV 3) (APR74648) |
| 2 | 1338/445 | Phosphoprotein (P) | Phosphoprotein (BYSMV 4) (AJW82843) |
| 3 | 570/189 | Movement protein (P3) | Gene 3 protein (NCMV 5) (NP_057956) |
| 4 | 237/79 | Hypothetical protein (P4) | No hit |
| 5 | 645/287 | Matrix protein (M) | No hit |
| 6 | 1560/520 | Glycoprotein (G) | Glycoprotein (YmCaV 6) (ARA91090) |
| 7 | 6342/2114 | RdRP 2 (L) | RdRp 2 (BYSMV 4) (YP_009177231) |
| GenBank Accession | Virus | N | P | P3 | M | G | L |
|---|---|---|---|---|---|---|---|
| KY366322 | YmCaV 1 | 27 | 20 | 25 | 21 | 23 | 33 |
| KX525586 | RSMV 2 | 30 | 24 | 23 | 17 | 22 | 35 |
| KT381973 | CBDaV 3 | 26 | 24 | 19 | 18 | 21 | 38 |
| AB030277 | NCMV 4 | 26 | 23 | 23 | 16 | 24 | 39 |
| KM213865 | BYSM 5 | 26 | 26 | 21 | 18 | 25 | 39 |
| KY884303 | MYSV 6 | 29 | 22 | 19 | 15 | 23 | 39 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alves-Freitas, D.M.T.; Pinheiro-Lima, B.; Faria, J.C.; Lacorte, C.; Ribeiro, S.G.; Melo, F.L. Double-Stranded RNA High-Throughput Sequencing Reveals a New Cytorhabdovirus in a Bean Golden Mosaic Virus-Resistant Common Bean Transgenic Line. Viruses 2019, 11, 90. https://doi.org/10.3390/v11010090
Alves-Freitas DMT, Pinheiro-Lima B, Faria JC, Lacorte C, Ribeiro SG, Melo FL. Double-Stranded RNA High-Throughput Sequencing Reveals a New Cytorhabdovirus in a Bean Golden Mosaic Virus-Resistant Common Bean Transgenic Line. Viruses. 2019; 11(1):90. https://doi.org/10.3390/v11010090
Chicago/Turabian StyleAlves-Freitas, Dione M. T., Bruna Pinheiro-Lima, Josias C. Faria, Cristiano Lacorte, Simone G. Ribeiro, and Fernando L. Melo. 2019. "Double-Stranded RNA High-Throughput Sequencing Reveals a New Cytorhabdovirus in a Bean Golden Mosaic Virus-Resistant Common Bean Transgenic Line" Viruses 11, no. 1: 90. https://doi.org/10.3390/v11010090
APA StyleAlves-Freitas, D. M. T., Pinheiro-Lima, B., Faria, J. C., Lacorte, C., Ribeiro, S. G., & Melo, F. L. (2019). Double-Stranded RNA High-Throughput Sequencing Reveals a New Cytorhabdovirus in a Bean Golden Mosaic Virus-Resistant Common Bean Transgenic Line. Viruses, 11(1), 90. https://doi.org/10.3390/v11010090