1. Introduction
Avian influenza A viruses are designated as either low pathogenicity avian influenza (LPAI) viruses or highly pathogenic avian influenza (HPAI) viruses based on their ability to cause severe disease and death in chickens in a laboratory setting. LPAI viruses are ubiquitous in wild birds throughout the world and HPAI viruses have evolved from subtype H5 and H7 LPAI viruses during replication in gallinaceous poultry. HPAI viruses have been reported in humans following direct or close contact with infected poultry, resulting in illness ranging from mild to severe. Mutations have been identified that appear to be necessary for avian influenza viruses to efficiently replicate in mammalian hosts, and it is now well documented that changes in the viral polymerase subunit PB2 (a key one being the E627K substitution [
1]) are necessary for this. This change has been identified in many avian subtype viruses, including H5N1 and H7N9 [
2,
3,
4,
5,
6,
7]. Other amino acid (AA) changes such as PB2 D701N (detected less commonly in human isolates of H5N1 and H7N7 [
8]), and the PB2 G590S, Q591R combination detected in A/H1N1pdm09 viruses [
9], similarly facilitate the adaptation of influenza viruses of non-human origin to humans. The PB2 protein is part of the influenza virus polymerase complex (comprising the PB2, PB1, PA subunits). The role of the PB2 protein is to bind to the methylated cap structures attached to host cell pre-mRNA segments, which are then cleaved by the PA protein and used by the PB1 protein to initiate transcription of viral mRNA segments. Increased nuclear entry of the PB2 protein via enhanced binding interactions between PB2 proteins and different isoforms of the mammalian importin-α protein, have been identified as the molecular mechanisms which enable PB2 proteins incorporating AA changes such as E627K and D701N to adapt to mammalian hosts [
10,
11,
12]. However, while many studies demonstrate the ability of avian influenza viruses to transition from low to high pathogenicity, these studies, which are most often performed in mice, usually involve serial passage in the mammalian host and do not provide information about when and how frequently the transition to the pathogenic phenotype occurs.
In order to understand the ability of avian influenza viruses to alter their pathogenic phenotype in a model better representing the human host, we infected naïve ferrets with an H5N1 avian influenza virus that initially demonstrated a low-pathogenic phenotype in ferrets. In addition to determining the mutations that gave rise to viruses with high pathogenicity for mammalian hosts, we investigated the rapidity and frequency of occurrence of such mutations and the location within the ferret organs where the mutated virus becomes established. These findings emphasize the potential of avian influenza viruses to select mutations that result in highly pathogenic infection, either in well documented virulence determinants or at additional sites, at disturbingly high frequency within a single passage in the ferret.
2. Materials and Methods
2.1. Cells and Viruses
African green monkey kidney (Vero) and Madin-Darby Canine Kidney (MDCK) cells were cultured in Eagle’s Minimal Essential Medium (EMEM, Thermo Fisher Scientific, Scoresby, Australia), supplemented with 10% (
v/
v) foetal calf serum (FCS, Thermo Fisher Scientific), 2 mM glutamine (Thermo Fisher Scientific), 10 mM
N-2-hydroxyethylpiperazine-
N′2-ethanesulfonic acid (HEPES, MP Biomedicals, Thermo Fisher Scientific), 100 units/mL penicillin (Sigma-Aldrich, Sydney, Australia), 100 µg/mL streptomycin (Sigma-Aldrich) and 2.5 µg/mL fungizone (Thermo Fisher Scientific). Human embryonic kidney-293T (293T) cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Thermo Fisher Scientific), plus supplements. Two H5N1 influenza A viruses, the human isolate A/Vietnam/1203/2004 (A/Viet, phylogenetic clade 1) [
13], and the avian virus A/Chicken/Laos/Xaythiani-26/2006 (A/Laos, clade 2.3) [
14] were obtained from the influenza virus repository at the AAHL. Both viruses have a multibasic cleavage site enabling replication in extra-respiratory tract sites and are highly pathogenic in chickens. Reverse-engineered viruses, denoted with the prefix rg (such as rgA/Laos), were constructed from RNA extracted and cloned from the A/Laos and A/Viet viruses using the eight-plasmid system [
15]. In preparation for inoculation into ferrets, all viruses were grown in the allantoic cavity of 9- to 11-day-old specific-pathogen-free embryonated hen’s eggs (Australian SPF services, Woodend, Australia) for up to 48 h. Harvested allantoic fluid was frozen at −80 °C and thawed immediately prior to use. The infectious titre of each stock was determined by titration in the allantoic cavity of embryonated eggs and the 50% egg infectious dose (EID
50)/mL was calculated according to the method of Reed and Muench [
16].
2.2. Ferret Experiments
Ferrets used in this study (sourced from IMVS, Adelaide, Australia) were 6–12 months old, of mixed gender, and all received food and water ad libitum while housed on a short light cycle (8 h light, 16 h dark), within the BSL-3 containment area at the AAHL. Prior to infection, a serum sample was taken from all ferrets and subjected to an influenza nucleoprotein (NP) competitive enzyme-linked immunosorbent assay (cELISA) capable of detecting antibodies against the influenza A virus nucleoprotein [
17], to confirm the seronegative nature of all ferrets to influenza A viruses. Ferret body weights were recorded, and nasal washes sampled immediately prior to infection and at regular intervals post infection (PI). On day 0 of each experiment, all ferrets were anaesthetised by intramuscular injection of 20 mg/mL Ilium Xylazil-20 (Troy-Laboratories, Glendenning, Australia) then intranasally inoculated with 0.5 mL PBS containing 10
6 EID
50 of virus (inoculum split between the two nares). On the days indicated in each experiment, nasal washes were sampled by aspirating 0.5 mL of PBS into each nare and collecting the ejected fluid which was stored at −80 °C until further use. During virus inoculation and nasal washing, ferrets were anaesthetised with ketamine/medetomidine (50:50, 1 mg/kg), reversed with atipemazole. In order to sample tissues on the days indicated in each experiment, ferrets were anaesthetised with ketamine as above and subsequently euthanised via intra-cardiac injection of pentabarbitone (150 mg/kg). Tissue samples (approximately 100 mg) were taken in sterile 2 mL tubes containing approximately 100 mg of silica-carbide chips (Daintree Scientific, St. Helens, Australia) and either 1 mL of PBS plus antibiotics for subsequent virus titrations, or 700 µL RLT buffer for subsequent isolation of vRNA. All tissue samples were homogenised twice at maximum speed for 30 s using a FastPrep-24 Instrument (MP biomedicals, Thermo Fisher Scientific) prior to storage at −80 °C.
All experiments involving ferrets were conducted with the approval of the CSIRO-AAHL Animal Ethics Committee (permit number 1280, approved 15/09/08). All procedures were conducted according to the guidelines of the Australian Government National Health and Medical Research Council as described in the Australian code for the care and use of animals for scientific purposes [
18].
2.3. Influenza NP cELISA
To detect broadly reactive influenza NP antibodies in ferret serum samples a competitive ELISA using plates coated with the NP from A/turkey/Ontario/6213/66 (H5N1) was used, as described by Selleck and Kirkland [
17]. Sera that inhibited 60% of the monoclonal antibody binding in this assay were considered antibody positive, while those that inhibited ≤40% of the monoclonal antibody binding were considered antibody negative.
2.4. Titration of Infectious Virus on Vero Cells
To determine the infectious virus titre, samples were titrated on Vero cells. Confluent Vero monolayers grown in 75 cm
2 flasks, were trypsinised and resuspended in 11 mL of EMEM plus supplements. For each 96-well plate, 1 mL of resuspended cells was added to 10 mL of EMEM plus supplements and 100 µL of this suspension loaded into each well of the plate. Nasal washes or 10% (
w/
v) tissue homogenates were serially diluted 10-fold in PBS and 100 µL aliquots were added to the 96-well plates freshly seeded with Vero cells. Plates were incubated at 37 °C, 5% (
v/
v) CO
2 in a humidified incubator for five days. On the 5th day all wells were visually assessed by light microscope for the presence of cytopathic effect (absence of a monolayer accompanied by dead cells) and the 50% tissue culture infectious dose (TCID
50) calculated according to the method of Reed and Muench [
16].
2.5. Immunohistochemical Analysis of Viral Antigen
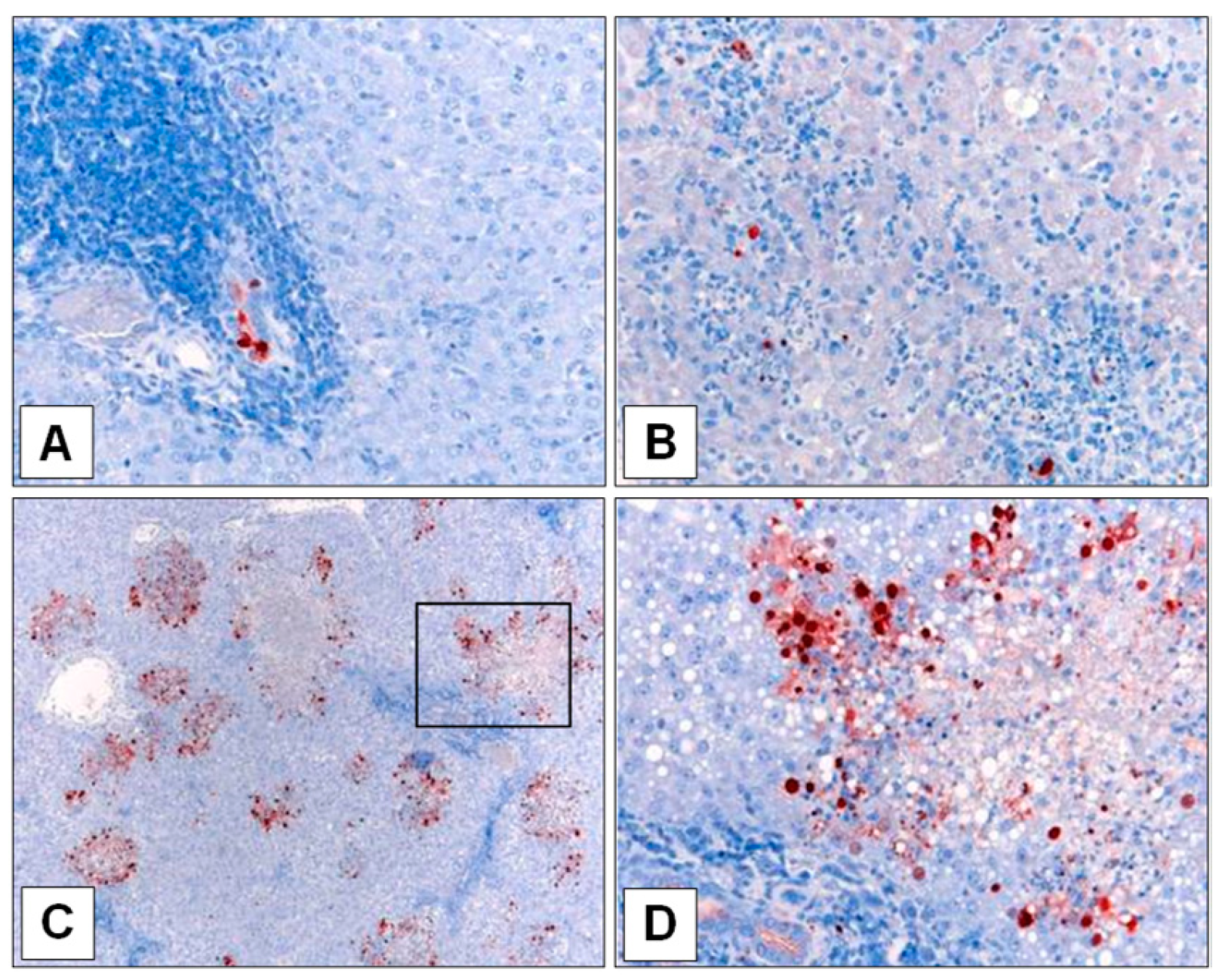
Following euthanasia, pieces of tissue ≤1 cm thick were fixed in neutral buffered formalin, embedded in paraffin and mounted on glass slides. Immunohistochemical staining of influenza viral antigen was performed using a rabbit polyclonal antiserum directed against a recombinant-expressed influenza viral NP (produced at the AAHL) and the DAKO EnVision® + System-HRP-(AEC) (Agilent Technologies, Mulgrave, Australia), according to the manufacturer’s instructions. Each section was then lightly counterstained for 2 min in Mayer’s haematoxylin solution (Lillie’s modification) prior to examination.
2.6. RNA Isolation, RT-PCR and Sequencing
RNA was extracted from allantoic fluid, nasal washes and animal tissues using an RNeasy Minikit (QIAGEN, Chadstone, Australia) following the manufacturer’s instructions. RT-PCR was performed in a one-step reaction using SuperScript One-Step RT-PCR for Long Templates (Thermo Fisher Scientific) and relevant primers matching the sequence of each viral genome segment (sequences available upon request). DNA sequencing was performed at the AAHL DNA sequencing facility, using an ABI PRISM 377 automated DNA sequencer (Thermo Fisher Scientific). DNA sequences obtained were translated and the deduced AA sequences aligned using Clone Manager v9.11 (Scientific & Educational Software, USA).
2.7. PB2 627 Single Nucleotide Polymorphism Analysis
A semi-quantitative PCR single nucleotide polymorphism (SNP) assay was developed to allow rapid discrimination between the nucleotide codons encoding the AA residues E and K at PB2 627 of A/Laos. Viral RNA was subjected to reverse transcription to generate viral cDNA using the Quantitect Reverse Transcription Kit (QIAGEN) according to the manufacturer’s instructions. The cDNA templates were subjected to SNP analysis using a set of primers and probes (sequences available upon request) and a Type-it Fast SNP Probe PCR kit (QIAGEN), according to the manufacturer’s instructions. Reactions were performed on an AB7900HT Real time PCR system (Thermo Fisher Scientific), held for 5 min at 95 °C and then subjected to 45 cycles of PCR consisting of denaturing at 95 °C for 15 s and a combined annealing and extension at 60 °C for 30 s, using fast cycling parameters. Following the completion of each test, cycle threshold (Ct) values were assessed using the Sequence Detection System software version 2.3 (Thermo Fisher Scientific). Only those samples that produced a characteristic exponential amplification curve resulting in a Ct value of ≤38 with either probe were considered to contain A/Laos virus encoding either PB2 627 E or K, or both.
2.8. Reverse-Engineered Viruses and Site-Directed Mutagenesis
All eight genome segments of the A/Laos and A/Viet H5N1 viruses were amplified by RT-PCR and incorporated into the pHW2000 reverse genetics (rg) virus rescue plasmid [
15]. Reverse-engineered viruses were generated by transfection of all eight plasmids into a co-culture of 293T and MDCK cells, as previously described [
15]. Supernatant that was haemagglutination positive was then amplified by a single virus passage in the allantoic cavity of 9- to 11-day-old embryonated hen’s eggs.
In order to investigate the effect of the PB2 627, 489 and NP 408 AA changes upon viral pathogenicity, plasmids encoding the A/Laos PB2 and NP as well as the A/Viet PB2 genome segments were subjected to site-directed mutagenesis using the GeneArt Site-Directed Mutagenesis System (Thermo Fisher Scientific) and relevant primer pairs (sequences available upon request). Following sequencing to confirm that the correct mutation had been achieved and that no other changes had been acquired, reverse-engineered viruses were generated by transfection of all eight plasmids into a co-culture of 293T and MDCK cells, as previously described [
15]. The correct PB2 and/or NP sequence was confirmed by DNA sequencing for all generated viruses.
2.9. In Vitro Analysis of Viral Polymerase Activity
The in vitro replication efficiency of different viral polymerase combinations was determined using 293T cells transfected with 10 µg each of plasmids encoding NP, PB1, PB2, PA, in conjunction with 0.5 µL of the pRL-TK plasmid (Promega, Alexandria, Australia), which encodes the renilla luciferase internal control protein, and 10 µg of a plasmid encoding the firefly luciferase protein surrounded by the 5′ and 3′ non-coding regions of the A/Viet NP vRNA segment (pPOL-NP-Luc). The pPOL-NP-Luc plasmid was first constructed by PCR amplification of the firefly luciferase gene from the pGL3 promoter vector plasmid (Promega) using relevant primers engineered to encode either the A/Viet NP 5′ or 3′ non-coding regions, in frame with a 5′ or 3′ segment of the luciferase gene. The resulting PCR product was cloned into the pPOL-I plasmid, containing a truncated human RNA polymerase I promoter and the hepatitis delta virus ribozyme (kindly donated by Glenn Marsh, AAHL), to generate pPOL-NP-Luc. Each plasmid mixture was made up to 100 µL with Opti-MEM (Thermo Fisher Scientific); 1 µL of Lipofectamine 2000 (Thermo Fisher Scientific) was added and the mixture incubated at room temperature for 20 min. A confluent monolayer of 293T cells grown in a 75 cm2 flask, was trypsinised, pelleted and resuspended in 10 mL DMEM plus supplements. A 200 µL aliquot of this suspension was then added to each well of a white sterile flat-bottomed 24-well plate. Each 100 µL minigenome complex was then added to a single well and the plates incubated at 37 °C, 5% (v/v) CO2 in a humidified incubator for 5 h, after which another 500 µL of DMEM plus supplements was added to each well and the incubation continued overnight. The next day the relative activity of each polymerase complex was assessed using the Dual-Luciferase Reporter Assay System (Promega), and a Fluoroskan Ascent FL 96-well plate reader (Thermo Fisher Scientific). The polymerase activity in each sample was normalised by dividing the amount of firefly luciferase detected in each sample by the amount of renilla luciferase detected in each sample. The resulting level of luciferase expression in the negative control sample, containing only plasmids pRL-TK and pPOL-NP-Luc was set at one unit, and the polymerase activity detected in each sample expressed as a fold increase relative to this control. The relative luciferase activities of the different polymerase complexes were compared by a one-way analysis of variance test using GraphPad Prism 4 for windows (GraphPad Software, Inc., San Diego, CA, USA).
4. Discussion
While similar studies of this type have been carried out previously [
21,
22], this study, performed in the ferret model, posed several important questions relating to the process of avian influenza adaption to the mammalian host that have yet to be fully considered in this model. We attempted to define how readily such adaption occurs, both in terms of frequency and rapidity. We also asked where in the animal do these viral adaptive mutations arise after intranasal infection and what are the implications of these mutations. Finally, we sought to understand the nature of the adapted virus.
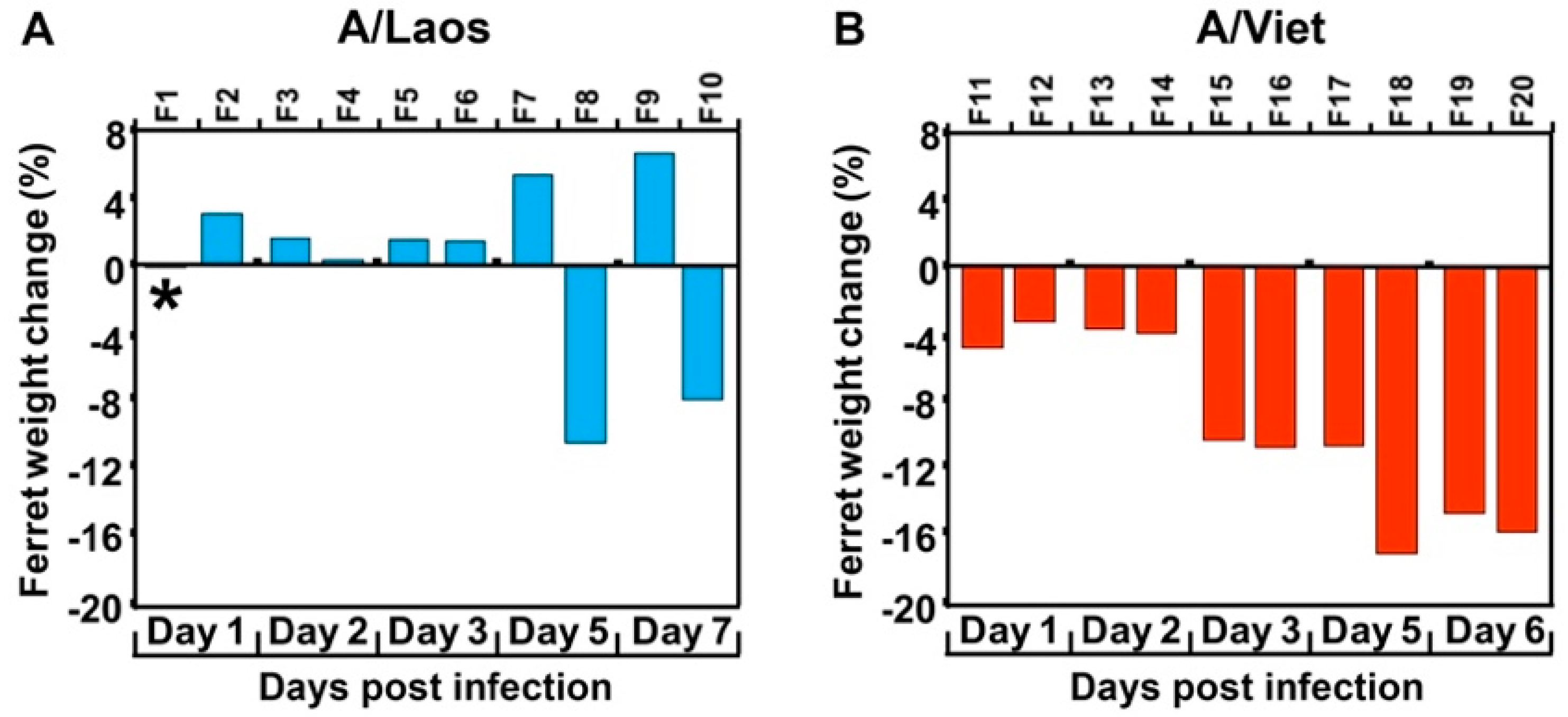
Use of the A/Laos isolate that was considered to be of low pathogenicity in mammals [
23], allowed for mammalian adaptation to be manifested by the detection of systemic spread by virtue of the multibasic cleavage site present in the HA of this virus. We showed that the pathogenic outcome was essentially binary; after infection of ferrets with the avian virus, the virus was either cleared from the respiratory tract by around 5 days, or the infection persisted in the upper respiratory tract beyond 5 days and became extra-respiratory. There were no cases of virus persisting beyond 5 days and not becoming extra-respiratory or of virus replicating in tissues outside the respiratory tract after being cleared from the respiratory tract. This is not to indicate that virus with adaptive mutations was not present in the animals until 5 days PI but that selection of such mutants and their rise to functional dominance took several days.
Another overarching observation was that the magnitude of virus loads in the nasal wash did not appear to predict systemic infection. Ferrets infected with A/Viet, all of which displayed systemic infection, had nasal wash titers ranging from 103.3–106.8, and those A/Laos-infected ferrets that ultimately showed viral replication outside the respiratory tract had nasal wash titers at both extremes of this range when systemic infection was detected. This is potentially related to the magnitude and effectiveness of innate defenses in individual animals. In terms of the frequency of adaptation, of the 20 ferrets sampled on days 5 or 7 PI with A/Laos virus or a virus inoculum (including reverse engineered virus) shown not to have adaptive mutations, eight of these (40%) acquired the capacity to grow in organs beyond the respiratory tract. Of these eight, all but one showed infectious virus in the small intestine and three showed infectious virus in the liver. Other sites were spleen, lymph node and olfactory bulb. Ferrets infected with virus already having adaptive mutations had a more extensive spread of infection starting from day 1 PI; of the 26 ferrets in this category (sampled from day 1–7 after infection with an adapted virus), the number of infected organs outside the respiratory tract ranged from one to seven (mean = 3.25). Those ferrets in which only one extra-respiratory organ was infected tended to be those sampled at the earlier time points (days 1–3) suggesting progressive seeding of virus throughout the animals with time. Interestingly, these early replication sites in ferrets infected with an already adapted virus, which were at disparate sites throughout the animals, did not correspond to the sites of first appearance of virus after infection with a non-adapted virus inoculum (small intestine, liver), perhaps suggesting that adaption is favoured in the small intestine or liver or that newly adapted virus more readily replicates in these sites.
When considering the implications of these findings for humans, as humans are infected with H5N1 viruses directly from an avian source and not currently from other infected humans, the majority will be expected to have a mild respiratory infection which is rapidly cleared and thus will probably go unreported. The unlucky and substantial percentage progressing to severe systemic disease are likely to experience a short initial lag phase in which viral replication may be inefficient before rapid expansion of the adapted virus in various organs, perhaps initially the small intestine or liver. Indeed, the incubation period in humans exposed to infected poultry has ranged from 2 to 10 days [
24], and the mean length of illness for fatal H5N1 infections in humans is 9 to 10 days [
25]. Furthermore, many reported cases of human H5N1 infection have been characterised by a period of illness apparently confined to either the gastrointestinal or respiratory tracts followed by a rapid systemic spread and subsequent death [
26,
27]. This clinical course of events reflects the observations made in ferrets during this study; failure to promptly clear the virus from the respiratory tract may provide an opportunity for the genetic changes that lead to a systemic infection to arise spontaneously due to errors in replication. This suggests that evidence of prolonged respiratory infection with H5N1 virus may serve as a useful prognostic indicator of systemic infection and highlights the need for prompt clearance of virus from the respiratory tract within the first 3 or 4 days in order to prevent the development of overwhelming systemic disease. Hence, the days immediately following a suspected infection likely represent a critical window of opportunity for the successful implementation of disease intervention strategies, such as the application of antiviral therapeutics, beyond which the challenge of successfully resolving a H5N1 infection presumably becomes increasingly difficult.
It should be noted that the viruses used in this study do not have the additional mutations in the HA identified by Imai et al. [
28] and Herfst et al. [
29] that are required for H5N1 viruses to become readily transmissible between ferrets by respiratory droplets, though it is understood that mutations in the polymerase genes that enhance viral replication in mammalian cells are an important step in this process [
9,
30,
31,
32,
33]. The most widely studied of these polymerase mutations is PB2 E627K, which is commonly identified in H5N1 and H7N9 viruses isolated from humans [
4,
21,
34]. The results of this study support the findings that the acquisition of the PB2 E627K mutation is the most common route to efficient replication in the mammalian host, though not the only one, and underscore the fact that such mutations can occur readily within a single passage in ferrets. The PB2 E627K mutation was initially described as being associated with overcoming cold-sensitivity [
35], improving replication in the nasal turbinates and in tissue culture at 33 °C [
35,
36]. It was also suggested that this mutation provided a platform for further mutations that may facilitate human-to-human transmission. If this was the case, we might expect to see a dominance of virus bearing PB2 627K in the nasal wash of all ferrets in which systemic spread had occurred. While this was usually the case, ferret F10 was an example where the virus in the liver and other organs had acquired PB2 627K, however virus from this animal that remained in the nasal wash encoded PB2 627E. It is difficult to envisage that virus containing PB2 627K could be shed from such an animal when only PB2 627E could be isolated from the upper respiratory tract. This was not to say that the nasal wash virus from ferret F10 was unadapted. In fact, the amplified virus was shown to contain two mutations, PB2 S489P and NP V408I, which were, like the PB2 E627K mutation [
37], able to enhance polymerase activity in mammalian cells when present in combination.
Although various combinations of mutations have been described in the literature for promoting mammalian adaptation [
38,
39,
40,
41], the PB2 S489P and NP V408I combination appears to be novel. The PB2 S489P single mutation has previously been described for its ability to enhance avian H5N1 polymerase activity in cultured human cells [
42]. It has also been detected in combination with the PB2 E627K mutation during experimental infection of mice with an H7N1 highly pathogenic avian influenza virus that developed enhanced pathogenicity during murine infection [
43]. Most combinations described to date involve the polymerase subunit genes [
38,
44,
45], but an adaptive combination of PB2 and NP changes, has been previously described by Danzy et al. [
46]. Interestingly, it was noted in their study that the NP mutation did not contribute to enhancing the polymerase activity which is contrary to our findings. During the course of our study we also identified other polymerase changes that occurred in the liver of ferret F8, namely PB2 I385V, PB2 N456D and PA E623G. However, the second ferret passage of virus from the F8 liver did not result in substantial weight loss and extra-respiratory tract replication was confined to the small intestine of two infected ferrets while the other two ferrets showed no systemic spread. It is tempting to suggest that these changes are in themselves insufficient to confer the pathogenic phenotype but may be intermediates for such pathogenic combinations to arise and reflect the dynamic nature of the influenza genome. The need for changes in both the PB2 and the NP for optimal replication efficiency are likely explained by the requirement of these proteins to acquire specificity for importin-α7 in mammalian cells as opposed to importin-α3, which avian viruses depend on for entry of the genome into the nucleus for replication [
12], or additionally for other barriers that have been described [
47] to be overcome. Perhaps the small intestine and liver, where we observe the adaptive process initially manifesting, provide sufficient levels of replication to increase the probability of a second mutation to occur.
The work presented in this study, using the ferret model, demonstrates that H5N1 viruses are capable of mutating to achieve high levels of replication in mammals by acquiring polymerase changes such as PB2 E627K or the PB2 S489P, NP V408I combination, rapidly and at high frequency. Furthermore, this process effectively converts an infection which is initially of low pathogenicity in the newly infected mammalian host to one of high pathogenicity in the course of a single virus passage in less than 5 days. These findings highlight the risks posed by ongoing human H5N1 infections where such mutations may form the foundation for further mutation towards a transmissible phenotype and reinforce the importance of preventing such infections.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




