Evasion of Innate and Intrinsic Antiviral Pathways by the Zika Virus

{kind=link}
Abstract
:1. Introduction
2. The Type I IFN System and Its Role in Restricting ZIKV Infection
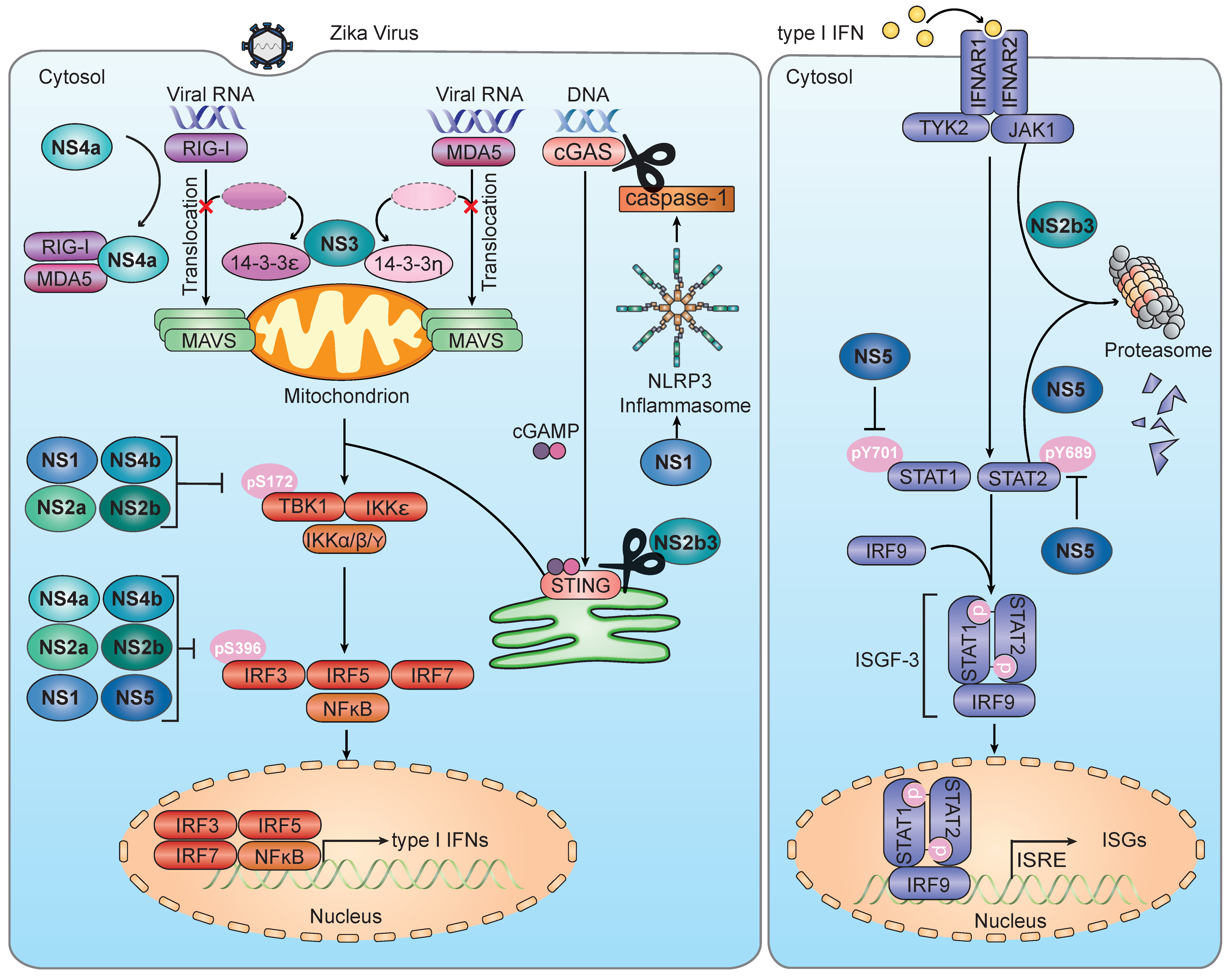
3. Innate Immune Sensing of Infection by ZIKV and Other Flaviviruses
3.1. TLR Sensing of ZIKV and Other Flaviviruses
3.2. RLR Sensing of ZIKV and Other Flaviviruses
3.3. cGAS-STING Sensing of ZIKV and Other Flaviviruses
4. Inhibition of IFN Induction by ZIKV and Other Flaviviruses
4.1. Mechanisms by Which DENV, WNV, and HCV Suppress IFN Production
4.2. Mechanisms by Which ZIKV Inhibits IFN Production
4.2.1. Evasion of RLR-MAVS Signaling
4.2.2. Evasion of the cGAS-STING Axis
4.2.3. Evasion of TBK1 and IRF3
5. Inhibition of IFNAR Signaling by ZIKV and Other Flaviviruses
5.1. Major Mechanisms by Which DENV, WNV and HCV Evade IFNAR Signaling
5.2. Evasion of the JAK–STAT Signaling Pathway by ZIKV
6. Manipulation of Other Antiviral Pathways by ZIKV
7. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Song, B.H.; Yun, S.I.; Woolley, M.; Lee, Y.M. Zika virus: History, epidemiology, transmission, and clinical presentation. J. Neuroimmunol. 2017, 308, 50–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, J.S.; Gubler, D.J.; Petersen, L.R. Emerging flaviviruses: The spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat. Med. 2004, 10, S98–S109. [Google Scholar] [CrossRef] [PubMed]
- Calvet, G.; Aguiar, R.S.; Melo, A.S.O.; Sampaio, S.A.; de Filippis, I.; Fabri, A.; Araujo, E.S.M.; de Sequeira, P.C.; de Mendonca, M.C.L.; de Oliveira, L.; et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: A case study. Lancet Infect. Dis. 2016, 16, 653–660. [Google Scholar] [CrossRef]
- McDonald, E.M.; Duggal, N.K.; Brault, A.C. Pathogenesis and sexual transmission of Spondweni and Zika viruses. PLoS Negl. Trop. Dis. 2017, 11, e0005990. [Google Scholar] [CrossRef] [PubMed]
- Foy, B.D.; Kobylinski, K.C.; Chilson Foy, J.L.; Blitvich, B.J.; Travassos da Rosa, A.; Haddow, A.D.; Lanciotti, R.S.; Tesh, R.B. Probable non-vector-borne transmission of Zika virus, Colorado, USA. Emerg. Infect. Dis. 2011, 17, 880–882. [Google Scholar] [CrossRef]
- Weaver, S.C.; Costa, F.; Garcia-Blanco, M.A.; Ko, A.I.; Ribeiro, G.S.; Saade, G.; Shi, P.Y.; Vasilakis, N. Zika virus: History, emergence, biology, and prospects for control. Antivir. Res. 2016, 130, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lormeau, V.M.; Blake, A.; Mons, S.; Lastere, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barre Syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef]
- Ventura, C.V.; Maia, M.; Dias, N.; Ventura, L.O.; Belfort, R., Jr. Zika: Neurological and ocular findings in infant without microcephaly. Lancet 2016, 387, 2502. [Google Scholar] [CrossRef]
- Driggers, R.W.; Ho, C.Y.; Korhonen, E.M.; Kuivanen, S.; Jaaskelainen, A.J.; Smura, T.; Rosenberg, A.; Hill, D.A.; DeBiasi, R.L.; Vezina, G.; et al. Zika Virus Infection with Prolonged Maternal Viremia and Fetal Brain Abnormalities. N. Engl. J. Med. 2016, 374, 2142–2151. [Google Scholar] [CrossRef]
- Mlakar, J.; Korva, M.; Tul, N.; Popovic, M.; Poljsak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodusek, V.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, M.S. The emergence of Zika virus and its new clinical syndromes. Nature 2018, 560, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Diamond, M.S. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe 2017, 21, 134–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Richard, A.S.; Shim, B.S.; Kwon, Y.C.; Zhang, R.; Otsuka, Y.; Schmitt, K.; Berri, F.; Diamond, M.S.; Choe, H. AXL-dependent infection of human fetal endothelial cells distinguishes Zika virus from other pathogenic flaviviruses. Proc. Natl. Acad. Sci. USA 2017, 114, 2024–2029. [Google Scholar] [CrossRef] [Green Version]
- Olagnier, D.; Muscolini, M.; Coyne, C.B.; Diamond, M.S.; Hiscott, J. Mechanisms of Zika Virus Infection and Neuropathogenesis. DNA Cell Biol. 2016, 35, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Kuhn, R.J.; Rossmann, M.G. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 2005, 3, 13–22. [Google Scholar] [CrossRef]
- Bollati, M.; Alvarez, K.; Assenberg, R.; Baronti, C.; Canard, B.; Cook, S.; Coutard, B.; Decroly, E.; de Lamballerie, X.; Gould, E.A.; et al. Structure and functionality in flavivirus NS-proteins: Perspectives for drug design. Antivir. Res. 2010, 87, 125–148. [Google Scholar] [CrossRef]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 2018, 16, 125–142. [Google Scholar] [CrossRef]
- Funk, A.; Truong, K.; Nagasaki, T.; Torres, S.; Floden, N.; Balmori Melian, E.; Edmonds, J.; Dong, H.; Shi, P.Y.; Khromykh, A.A. RNA structures required for production of subgenomic flavivirus RNA. J. Virol. 2010, 84, 11407–11417. [Google Scholar] [CrossRef]
- Pijlman, G.P.; Funk, A.; Kondratieva, N.; Leung, J.; Torres, S.; van der Aa, L.; Liu, W.J.; Palmenberg, A.C.; Shi, P.Y.; Hall, R.A.; et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 2008, 4, 579–591. [Google Scholar] [CrossRef]
- Goertz, G.P.; Abbo, S.R.; Fros, J.J.; Pijlman, G.P. Functional RNA during Zika virus infection. Virus Res. 2018, 254, 41–53. [Google Scholar] [CrossRef] [PubMed]
- MacFadden, A.; O’Donoghue, Z.; Silva, P.; Chapman, E.G.; Olsthoorn, R.C.; Sterken, M.G.; Pijlman, G.P.; Bredenbeek, P.J.; Kieft, J.S. Mechanism and structural diversity of exoribonuclease-resistant RNA structures in flaviviral RNAs. Nat. Commun. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Mietchen, D.; Coffee, M.; Stratton, T.P.; Freundlich, J.S.; Freitas-Junior, L.; Muratov, E.; Siqueira-Neto, J.; Williams, A.J.; Andrade, C. Open drug discovery for the Zika virus. F1000Research 2016, 5, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, H.; Ji, X.; Yang, X.; Xie, W.; Yang, K.; Chen, C.; Wu, C.; Chi, H.; Mu, Z.; Wang, Z.; et al. The crystal structure of Zika virus helicase: Basis for antiviral drug design. Protein Cell 2016, 7, 450–454. [Google Scholar] [CrossRef]
- Wang, B.; Thurmond, S.; Hai, R.; Song, J. Structure and function of Zika virus NS5 protein: Perspectives for drug design. Cell. Mol. Life Sci. 2018, 75, 1723–1736. [Google Scholar] [CrossRef]
- Wang, Q.; Yan, J.; Gao, G.F. Monoclonal Antibodies against Zika Virus: Therapeutics and Their Implications for Vaccine Design. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [Green Version]
- Bowen, J.R.; Quicke, K.M.; Maddur, M.S.; O’Neal, J.T.; McDonald, C.E.; Fedorova, N.B.; Puri, V.; Shabman, R.S.; Pulendran, B.; Suthar, M.S. Zika Virus Antagonizes Type I Interferon Responses during Infection of Human Dendritic Cells. PLoS Pathog. 2017, 13, e1006164. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aliota, M.T.; Caine, E.A.; Walker, E.C.; Larkin, K.E.; Camacho, E.; Osorio, J.E. Characterization of Lethal Zika Virus Infection in AG129 Mice. PLoS Negl. Trop. Dis. 2016, 10, e0004682. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Balasubramaniam, V.R.; Brown, J.A.; Mena, I.; Grant, A.; Bardina, S.V.; Maringer, K.; Schwarz, M.C.; Maestre, A.M.; Sourisseau, M.; et al. A novel Zika virus mouse model reveals strain specific differences in virus pathogenesis and host inflammatory immune responses. PLoS Pathog. 2017, 13, e1006258. [Google Scholar] [CrossRef] [PubMed]
- Yockey, L.J.; Jurado, K.A.; Arora, N.; Millet, A.; Rakib, T.; Milano, K.M.; Hastings, A.K.; Fikrig, E.; Kong, Y.; Horvath, T.L.; et al. Type I interferons instigate fetal demise after Zika virus infection. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Yockey, L.J.; Varela, L.; Rakib, T.; Khoury-Hanold, W.; Fink, S.L.; Stutz, B.; Szigeti-Buck, K.; Van den Pol, A.; Lindenbach, B.D.; Horvath, T.L.; et al. Vaginal Exposure to Zika Virus during Pregnancy Leads to Fetal Brain Infection. Cell 2016, 166, 1247–1256.e4. [Google Scholar] [CrossRef] [Green Version]
- Hertzog, J.; Dias Junior, A.G.; Rigby, R.E.; Donald, C.L.; Mayer, A.; Sezgin, E.; Song, C.; Jin, B.; Hublitz, P.; Eggeling, C.; et al. Infection with a Brazilian isolate of Zika virus generates RIG-I stimulatory RNA and the viral NS5 protein blocks type I IFN induction and signaling. Eur. J. Immunol. 2018, 48, 1120–1136. [Google Scholar] [CrossRef]
- Savidis, G.; Perreira, J.M.; Portmann, J.M.; Meraner, P.; Guo, Z.; Green, S.; Brass, A.L. The IFITMs Inhibit Zika Virus Replication. Cell Rep. 2016, 15, 2323–2330. [Google Scholar] [CrossRef] [Green Version]
- Sabouri, A.H.; Marcondes, M.C.; Flynn, C.; Berger, M.; Xiao, N.; Fox, H.S.; Sarvetnick, N.E. TLR signaling controls lethal encephalitis in WNV-infected brain. Brain Res. 2014, 1574, 84–95. [Google Scholar] [CrossRef] [Green Version]
- Suthar, M.S.; Aguirre, S.; Fernandez-Sesma, A. Innate immune sensing of flaviviruses. PLoS Pathog. 2013, 9, e1003541. [Google Scholar] [CrossRef]
- Sun, B.; Sundstrom, K.B.; Chew, J.J.; Bist, P.; Gan, E.S.; Tan, H.C.; Goh, K.C.; Chawla, T.; Tang, C.K.; Ooi, E.E. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci. Rep. 2017, 7, 3594. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, S.; Luthra, P.; Sanchez-Aparicio, M.T.; Maestre, A.M.; Patel, J.; Lamothe, F.; Fredericks, A.C.; Tripathi, S.; Zhu, T.; Pintado-Silva, J.; et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat. Microbiol. 2017, 2, 17037. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Chiang, J.J.; Davis, M.E.; Gack, M.U. Regulation of RIG-I-like receptor signaling by host and viral proteins. Cytokine Growth Factor Rev. 2014, 25, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Chiu, Y.H.; Chen, Z.J. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Suthar, M.S.; Diamond, M.S.; Gale, M., Jr. West Nile virus infection and immunity. Nat. Rev. Microbiol. 2013, 11, 115–128. [Google Scholar] [CrossRef]
- Town, T.; Bai, F.; Wang, T.; Kaplan, A.T.; Qian, F.; Montgomery, R.R.; Anderson, J.F.; Flavell, R.A.; Fikrig, E. Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity 2009, 30, 242–253. [Google Scholar] [CrossRef]
- Szretter, K.J.; Daffis, S.; Patel, J.; Suthar, M.S.; Klein, R.S.; Gale, M., Jr.; Diamond, M.S. The innate immune adaptor molecule MyD88 restricts West Nile virus replication and spread in neurons of the central nervous system. J. Virol. 2010, 84, 12125–12138. [Google Scholar] [CrossRef]
- Tsai, Y.T.; Chang, S.Y.; Lee, C.N.; Kao, C.L. Human TLR3 recognizes dengue virus and modulates viral replication in vitro. Cell. Microbiol. 2009, 11, 604–615. [Google Scholar] [CrossRef]
- Nasirudeen, A.M.; Wong, H.H.; Thien, P.; Xu, S.; Lam, K.P.; Liu, D.X. RIG-I, MDA5 and TLR3 synergistically play an important role in restriction of dengue virus infection. PLoS Negl. Trop. Dis. 2011, 5, e926. [Google Scholar] [CrossRef] [PubMed]
- Vanwalscappel, B.; Tada, T.; Landau, N.R. Toll-like receptor agonist R848 blocks Zika virus replication by inducing the antiviral protein viperin. Virology 2018, 522, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Fredericksen, B.L.; Keller, B.C.; Fornek, J.; Katze, M.G.; Gale, M., Jr. Establishment and maintenance of the innate antiviral response to West Nile Virus involves both RIG-I and MDA5 signaling through IPS-1. J. Virol. 2008, 82, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Suthar, M.S.; Ma, D.Y.; Thomas, S.; Lund, J.M.; Zhang, N.; Daffis, S.; Rudensky, A.Y.; Bevan, M.J.; Clark, E.A.; Kaja, M.K.; et al. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 2010, 6, e1000757. [Google Scholar] [CrossRef]
- Perry, S.T.; Prestwood, T.R.; Lada, S.M.; Benedict, C.A.; Shresta, S. Cardif-mediated signaling controls the initial innate response to dengue virus in vivo. J. Virol. 2009, 83, 8276–8281. [Google Scholar] [CrossRef]
- Ma, J.; Ketkar, H.; Geng, T.; Lo, E.; Wang, L.; Xi, J.; Sun, Q.; Zhu, Z.; Cui, Y.; Yang, L.; et al. Zika Virus Non-structural Protein 4A Blocks the RLR-MAVS Signaling. Front. Microbiol. 2018, 9, 1350. [Google Scholar] [CrossRef] [Green Version]
- Riedl, W.; Acharya, D.; Lee, J.H.; Liu, G.; Serman, T.; Chiang, C.; Chan, Y.K.; Diamond, M.S.; Gack, M.U. Zika Virus NS3 Mimics a Cellular 14-3-3-Binding Motif to Antagonize RIG-I- and MDA5-Mediated Innate Immunity. Cell Host Microbe 2019, 26, 493–503.e6. [Google Scholar] [CrossRef]
- Esser-Nobis, K.; Aarreberg, L.D.; Roby, J.A.; Fairgrieve, M.R.; Green, R.; Gale, M., Jr. Comparative Analysis of African and Asian Lineage-Derived Zika Virus Strains Reveals Differences in Activation of and Sensitivity to Antiviral Innate Immunity. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, Q.; Wu, Y.; Ma, L.; Zhang, Z.; Liu, T.; Jin, S.; She, Y.; Li, Y.P.; Cui, J. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Ding, Q.; Gaska, J.M.; Douam, F.; Wei, L.; Kim, D.; Balev, M.; Heller, B.; Ploss, A. Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proc. Natl. Acad. Sci. USA 2018, 115, E6310–E6318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gack, M.U.; Diamond, M.S. Innate immune escape by Dengue and West Nile viruses. Curr. Opin. Virol. 2016, 20, 119–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Kinch, L.N.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36, 959–973. [Google Scholar] [CrossRef]
- Manokaran, G.; Finol, E.; Wang, C.; Gunaratne, J.; Bahl, J.; Ong, E.Z.; Tan, H.C.; Sessions, O.M.; Ward, A.M.; Gubler, D.J.; et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 2015, 350, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 479–480, 110–121. [Google Scholar] [CrossRef]
- Liu, H.M.; Loo, Y.M.; Horner, S.M.; Zornetzer, G.A.; Katze, M.G.; Gale, M., Jr. The mitochondrial targeting chaperone 14-3-3epsilon regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe 2012, 11, 528–537. [Google Scholar] [CrossRef]
- Chan, Y.K.; Gack, M.U. A phosphomimetic-based mechanism of dengue virus to antagonize innate immunity. Nat. Immunol. 2016, 17, 523–530. [Google Scholar] [CrossRef]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [Green Version]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- He, Z.; Zhu, X.; Wen, W.; Yuan, J.; Hu, Y.; Chen, J.; An, S.; Dong, X.; Lin, C.; Yu, J.; et al. Dengue Virus Subverts Host Innate Immunity by Targeting Adaptor Protein MAVS. J. Virol. 2016, 90, 7219–7230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuessler, A.; Funk, A.; Lazear, H.M.; Cooper, D.A.; Torres, S.; Daffis, S.; Jha, B.K.; Kumagai, Y.; Takeuchi, O.; Hertzog, P.; et al. West Nile virus noncoding subgenomic RNA contributes to viral evasion of the type I interferon-mediated antiviral response. J. Virol. 2012, 86, 5708–5718. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.Y.; Hsu, T.W.; Chen, Y.L.; Liu, S.F.; Tsai, Y.J.; Lin, Y.T.; Chen, Y.S.; Fan, Y.H. Japanese encephalitis virus non-coding RNA inhibits activation of interferon by blocking nuclear translocation of interferon regulatory factor 3. Vet. Microbiol. 2013, 166, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, S.; Maestre, A.M.; Pagni, S.; Patel, J.R.; Savage, T.; Gutman, D.; Maringer, K.; Bernal-Rubio, D.; Shabman, R.S.; Simon, V.; et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 2012, 8, e1002934. [Google Scholar] [CrossRef]
- Yu, C.Y.; Chang, T.H.; Liang, J.J.; Chiang, R.L.; Lee, Y.L.; Liao, C.L.; Lin, Y.L. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog. 2012, 8, e1002780. [Google Scholar] [CrossRef]
- Ding, Q.; Cao, X.; Lu, J.; Huang, B.; Liu, Y.J.; Kato, N.; Shu, H.B.; Zhong, J. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J. Hepatol. 2013, 59, 52–58. [Google Scholar] [CrossRef]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, Q.; Zhou, J.; Xie, W.; Chen, C.; Wang, Z.; Yang, H.; Cui, J. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov. 2017, 3, 17006. [Google Scholar] [CrossRef]
- Xia, H.; Luo, H.; Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Medeiros, D.B.A.; Zou, J.; Xie, X.; Giraldo, M.I.; Vasconcelos, P.F.C.; et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat. Commun. 2018, 9, 414. [Google Scholar] [CrossRef]
- Lin, J.P.; Fan, Y.K.; Liu, H.M. The 14-3-3eta chaperone protein promotes antiviral innate immunity via facilitating MDA5 oligomerization and intracellular redistribution. PLoS Pathog. 2019, 15, e1007582. [Google Scholar] [CrossRef]
- Lin, S.; Yang, S.; He, J.; Guest, J.D.; Ma, Z.; Yang, L.; Pierce, B.G.; Tang, Q.; Zhang, Y.J. Zika virus NS5 protein antagonizes type I interferon production via blocking TBK1 activation. Virology 2019, 527, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, B.M.; Laurence, H.M.; Massey, A.R.; Costantino, D.A.; Xie, X.; Yang, Y.; Shi, P.Y.; Nix, J.C.; Beckham, J.D.; Kieft, J.S. Zika virus produces noncoding RNAs using a multi-pseudoknot structure that confounds a cellular exonuclease. Science 2016, 354, 1148–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donald, C.L.; Brennan, B.; Cumberworth, S.L.; Rezelj, V.V.; Clark, J.J.; Cordeiro, M.T.; Freitas de Oliveira Franca, R.; Pena, L.J.; Wilkie, G.S.; Da Silva Filipe, A.; et al. Full Genome Sequence and sfRNA Interferon Antagonist Activity of Zika Virus from Recife, Brazil. PLoS Negl. Trop. Dis. 2016, 10, e0005048. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.; Lennemann, N.J.; Ouyang, Y.; Bramley, J.C.; Morosky, S.; Marques, E.T., Jr.; Cherry, S.; Sadovsky, Y.; Coyne, C.B. Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus Infection. Cell Host Microbe 2016, 19, 705–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Zagorska, A.; Lew, E.D.; Shrestha, B.; Rothlin, C.V.; Naughton, J.; Diamond, M.S.; Lemke, G.; Young, J.A. Enveloped viruses disable innate immune responses in dendritic cells by direct activation of TAM receptors. Cell Host Microbe 2013, 14, 136–147. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef]
- Guo, J.T.; Hayashi, J.; Seeger, C. West Nile virus inhibits the signal transduction pathway of alpha interferon. J. Virol. 2005, 79, 1343–1350. [Google Scholar] [CrossRef]
- Liu, W.J.; Wang, X.J.; Mokhonov, V.V.; Shi, P.Y.; Randall, R.; Khromykh, A.A. Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins. J. Virol. 2005, 79, 1934–1942. [Google Scholar] [CrossRef]
- Munoz-Jordan, J.L.; Sanchez-Burgos, G.G.; Laurent-Rolle, M.; Garcia-Sastre, A. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA 2003, 100, 14333–14338. [Google Scholar] [CrossRef] [Green Version]
- Kumthip, K.; Chusri, P.; Jilg, N.; Zhao, L.; Fusco, D.N.; Zhao, H.; Goto, K.; Cheng, D.; Schaefer, E.A.; Zhang, L.; et al. Hepatitis C virus NS5A disrupts STAT1 phosphorylation and suppresses type I interferon signaling. J. Virol. 2012, 86, 8581–8591. [Google Scholar] [CrossRef]
- Laurent-Rolle, M.; Boer, E.F.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; et al. The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, M.; Jones, M.; Davidson, A.; Chain, B.; Jacobs, M. Dengue virus NS5 inhibits interferon-alpha signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J. Infect. Dis. 2009, 200, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.Y.; Garcia-Sastre, A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.; Laurent-Rolle, M.; Maestre, A.M.; Rajsbaum, R.; Pisanelli, G.; Simon, V.; Mulder, L.C.; Fernandez-Sesma, A.; Garcia-Sastre, A. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog. 2013, 9, e1003265. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Rolle, M.; Morrison, J.; Rajsbaum, R.; Macleod, J.M.L.; Pisanelli, G.; Pham, A.; Ayllon, J.; Miorin, L.; Martinez, C.; tenOever, B.R.; et al. The interferon signaling antagonist function of yellow fever virus NS5 protein is activated by type I interferon. Cell Host Microbe 2014, 16, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sanchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef]
- Van der Hoek, K.H.; Eyre, N.S.; Shue, B.; Khantisitthiporn, O.; Glab-Ampi, K.; Carr, J.M.; Gartner, M.J.; Jolly, L.A.; Thomas, P.Q.; Adikusuma, F.; et al. Viperin is an important host restriction factor in control of Zika virus infection. Sci. Rep. 2017, 7, 4475. [Google Scholar] [CrossRef]
- Joyce, M.A.; Berry-Wynne, K.M.; Dos Santos, T.; Addison, W.R.; McFarlane, N.; Hobman, T.; Tyrrell, D.L. HCV and flaviviruses hijack cellular mechanisms for nuclear STAT2 degradation: Up-regulation of PDLIM2 suppresses the innate immune response. PLoS Pathog. 2019, 15, e1007949. [Google Scholar] [CrossRef]
- Strange, D.P.; Jiyarom, B.; Pourhabibi Zarandi, N.; Xie, X.; Baker, C.; Sadri-Ardekani, H.; Shi, P.Y.; Verma, S. Axl Promotes Zika Virus Entry and Modulates the Antiviral State of Human Sertoli Cells. MBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Montero, H.; Trujillo-Alonso, V. Stress granules in the viral replication cycle. Viruses 2011, 3, 2328–2338. [Google Scholar] [CrossRef] [PubMed]
- Pager, C.T.; Schutz, S.; Abraham, T.M.; Luo, G.; Sarnow, P. Modulation of hepatitis C virus RNA abundance and virus release by dispersion of processing bodies and enrichment of stress granules. Virology 2013, 435, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Emara, M.M.; Brinton, M.A. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 9041–9046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonenfant, G.; Williams, N.; Netzband, R.; Schwarz, M.C.; Evans, M.J.; Pager, C.T. Zika Virus Subverts Stress Granules To Promote and Restrict Viral Gene Expression. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Kumar, A.; Xu, Z.; Airo, A.M.; Stryapunina, I.; Wong, C.P.; Branton, W.; Tchesnokov, E.; Gotte, M.; Power, C.; et al. Zika Virus Hijacks Stress Granule Proteins and Modulates the Host Stress Response. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Amorim, R.; Temzi, A.; Griffin, B.D.; Mouland, A.J. Zika virus inhibits eIF2alpha-dependent stress granule assembly. PLoS Negl. Trop. Dis. 2017, 11, e0005775. [Google Scholar] [CrossRef]
- Hug, N.; Longman, D.; Caceres, J.F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016, 44, 1483–1495. [Google Scholar] [CrossRef] [Green Version]
- Balistreri, G.; Horvath, P.; Schweingruber, C.; Zund, D.; McInerney, G.; Merits, A.; Muhlemann, O.; Azzalin, C.; Helenius, A. The host nonsense-mediated mRNA decay pathway restricts Mammalian RNA virus replication. Cell Host Microbe 2014, 16, 403–411. [Google Scholar] [CrossRef]
- Serquina, A.K.; Das, S.R.; Popova, E.; Ojelabi, O.A.; Roy, C.K.; Gottlinger, H.G. UPF1 is crucial for the infectivity of human immunodeficiency virus type 1 progeny virions. J. Virol. 2013, 87, 8853–8861. [Google Scholar] [CrossRef]
- Balistreri, G.; Bognanni, C.; Muhlemann, O. Virus Escape and Manipulation of Cellular Nonsense-Mediated mRNA Decay. Viruses 2017, 9. [Google Scholar] [CrossRef]
- Ramage, H.R.; Kumar, G.R.; Verschueren, E.; Johnson, J.R.; Von Dollen, J.; Johnson, T.; Newton, B.; Shah, P.; Horner, J.; Krogan, N.J.; et al. A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol. Cell 2015, 57, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.A.; Leon, K.E.; Khalid, M.M.; Tomar, S.; Jimenez-Morales, D.; Dunlap, M.; Kaye, J.A.; Shah, P.S.; Finkbeiner, S.; Krogan, N.J.; et al. The Cellular NMD Pathway Restricts Zika Virus Infection and Is Targeted by the Viral Capsid Protein. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Johnson, J.R.; Truong, B.; Kim, G.; Weinbren, N.; Dittmar, M.; Shah, P.S.; Von Dollen, J.; Newton, B.W.; Jang, G.M.; et al. Identification of antiviral roles for the exon-junction complex and nonsense-mediated decay in flaviviral infection. Nat. Microbiol. 2019, 4, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef]
- Ke, P.Y. The Multifaceted Roles of Autophagy in Flavivirus-Host Interactions. Int. J. Mol. Sci. 2018, 19, 3940. [Google Scholar] [CrossRef]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.S.; Lee, S.A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Liu, B.; Yves, T.D.; He, Y.; Wang, S.; Tang, H.; Ren, H.; Zhao, P.; Qi, Z.; Qin, Z. Zika Virus Induces Autophagy in Human Umbilical Vein Endothelial Cells. Viruses 2018, 10. [Google Scholar] [CrossRef]
- Cao, B.; Parnell, L.A.; Diamond, M.S.; Mysorekar, I.U. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J. Exp. Med. 2017, 214, 2303–2313. [Google Scholar] [CrossRef]
- Lennemann, N.J.; Coyne, C.B. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy 2017, 13, 322–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatogawa, H.; Mochida, K. Reticulophagy and nucleophagy: New findings and unsolved issues. Autophagy 2015, 11, 2377–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serman, T.M.; Gack, M.U. Evasion of Innate and Intrinsic Antiviral Pathways by the Zika Virus. Viruses 2019, 11, 970. https://doi.org/10.3390/v11100970
Serman TM, Gack MU. Evasion of Innate and Intrinsic Antiviral Pathways by the Zika Virus. Viruses. 2019; 11(10):970. https://doi.org/10.3390/v11100970
Chicago/Turabian StyleSerman, Taryn M., and Michaela U. Gack. 2019. "Evasion of Innate and Intrinsic Antiviral Pathways by the Zika Virus" Viruses 11, no. 10: 970. https://doi.org/10.3390/v11100970
APA StyleSerman, T. M., & Gack, M. U. (2019). Evasion of Innate and Intrinsic Antiviral Pathways by the Zika Virus. Viruses, 11(10), 970. https://doi.org/10.3390/v11100970