High Diversity and Novel Enteric Viruses in Fecal Viromes of Healthy Wild and Captive Thai Cynomolgus Macaques (Macaca fascicularis)


 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. Specimen Collection
2.3. Viral Enrichment and Extraction
2.4. Reverse Transcription and Random Amplification of Viral Genome
2.5. Virome Library Preparation and Sequencing
2.6. Data Processing and Analysis
2.7. Genome Acquisition of Novel Picornavirus
2.8. Genome Completion of The Novel Parvovirus
2.9. Phylogenetic Analysis
3. Results
3.1. The Study Population
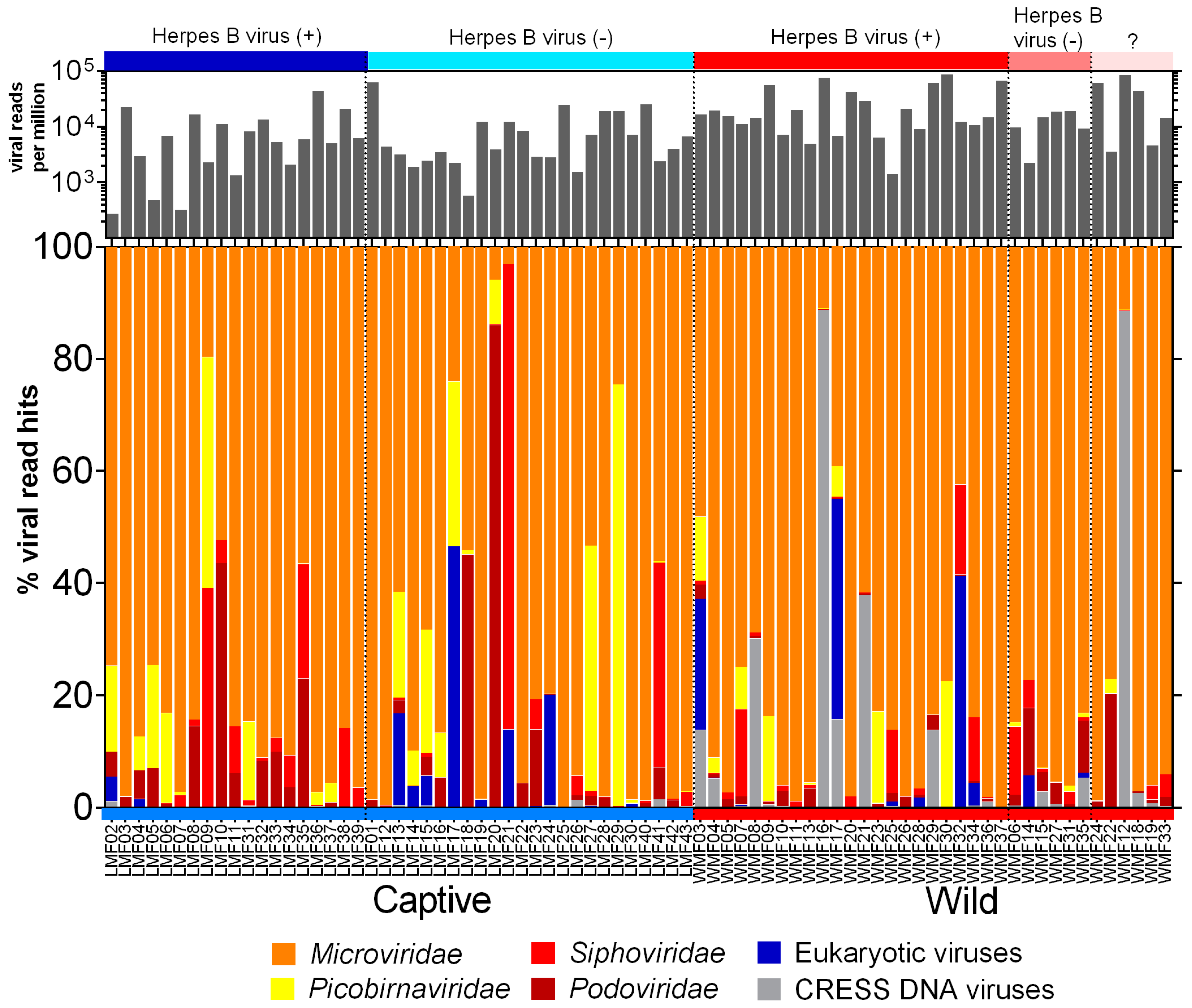
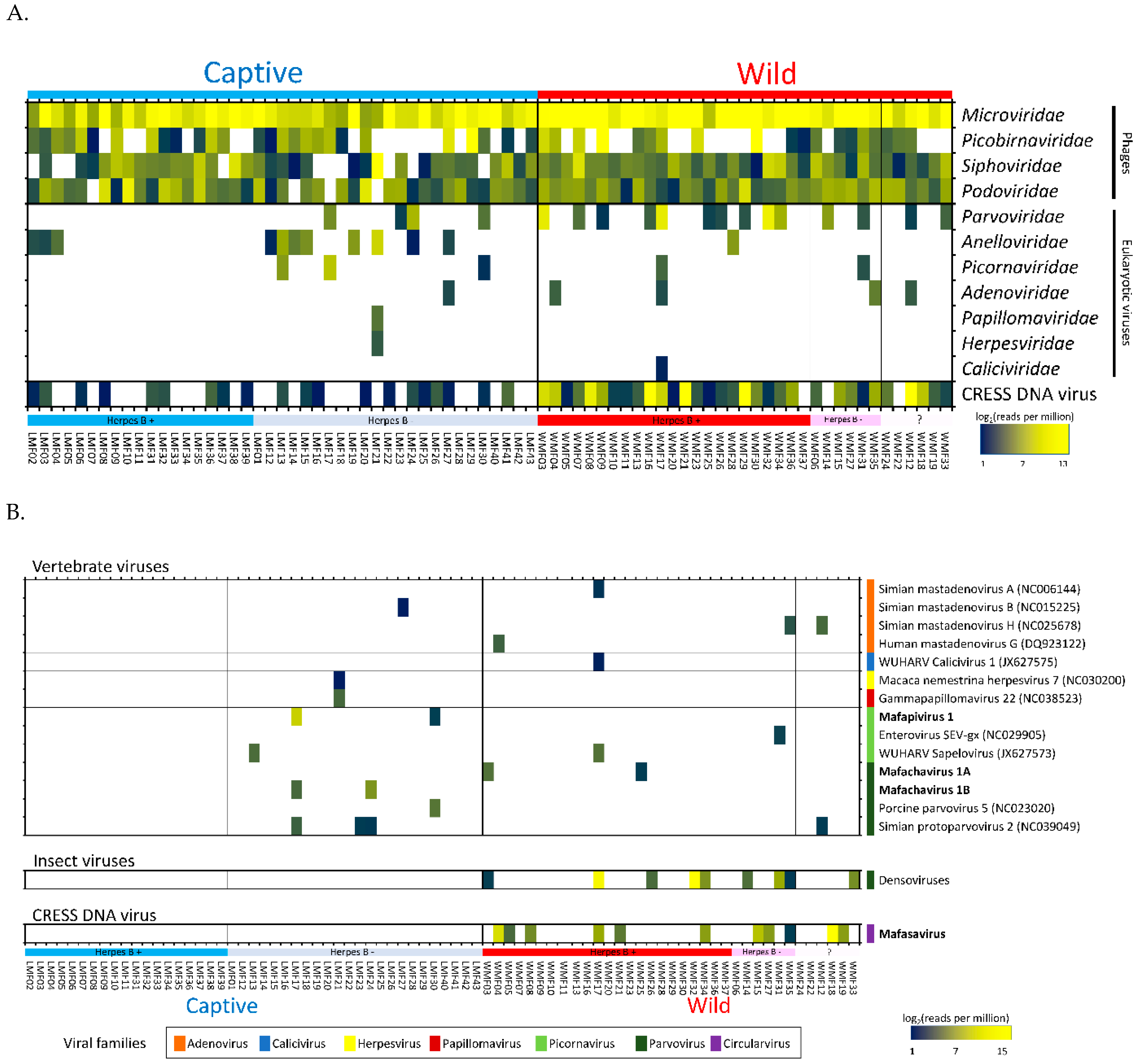
3.2. Virome Composition
3.3. Eukaryotic Viruses
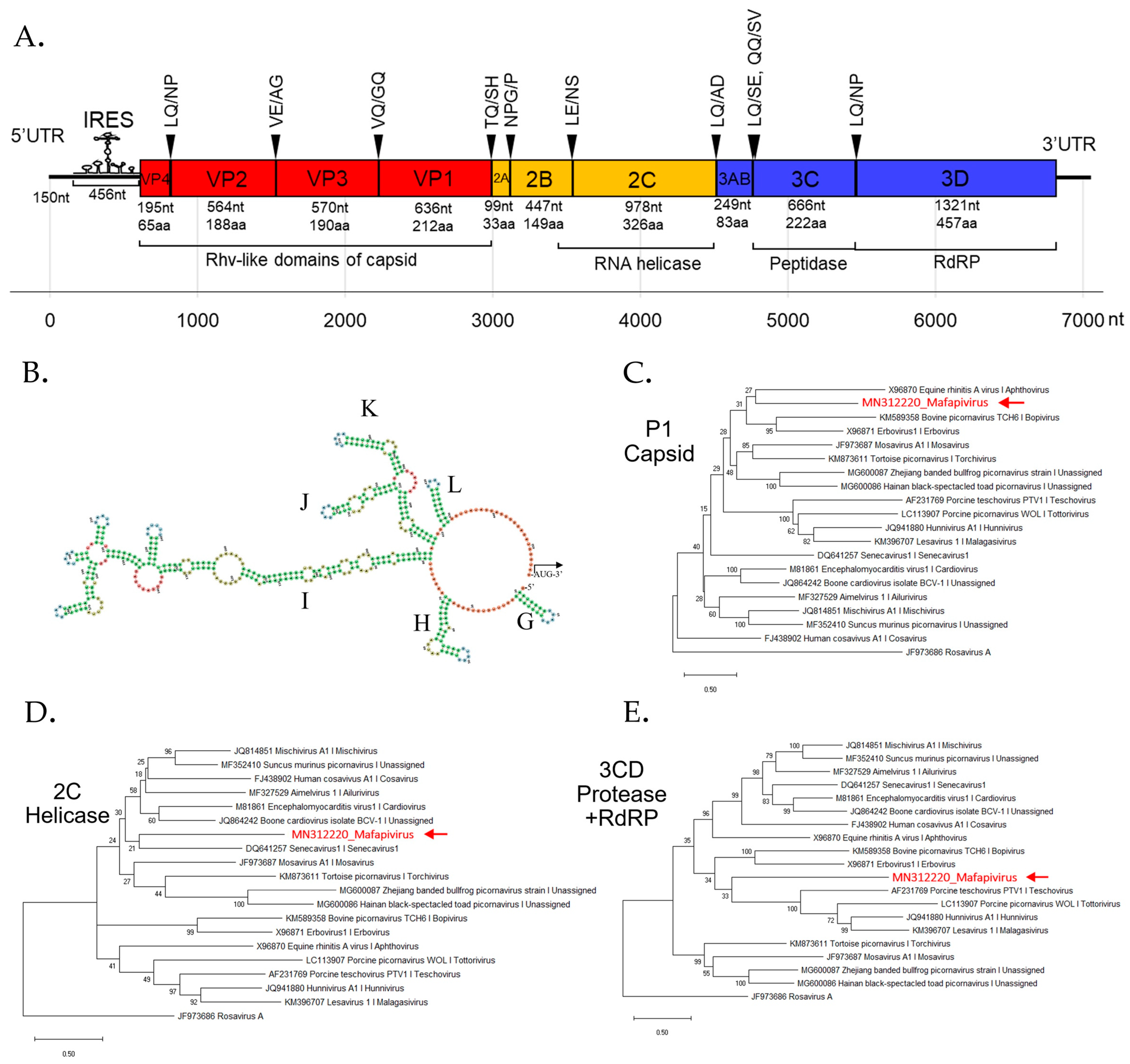
3.4. Detection of Three Picornaviruses
New Picornaviridae Genus
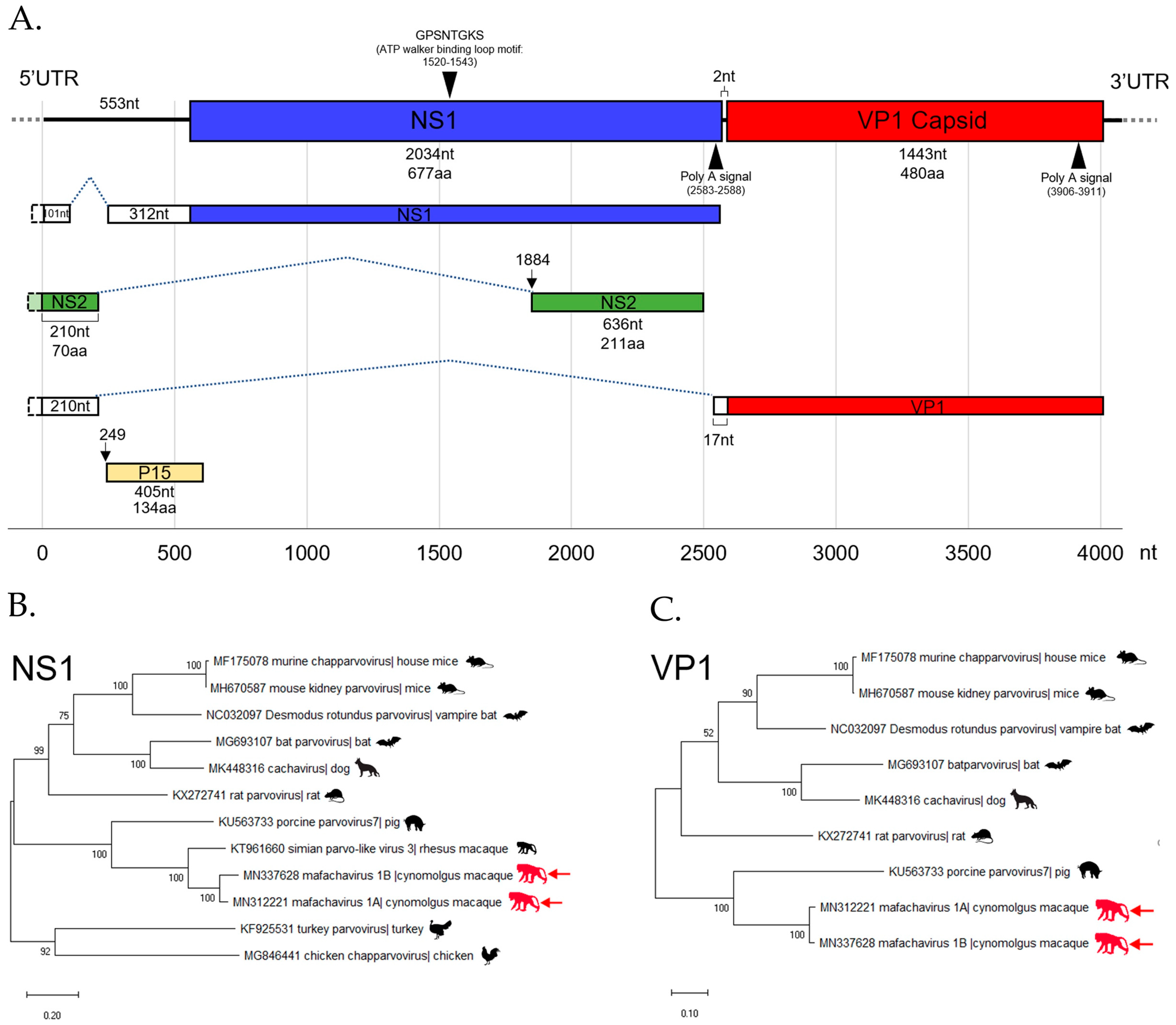
3.5. Parvovirus
The First Simian Chapparvovirus Genome with Complete ORFs
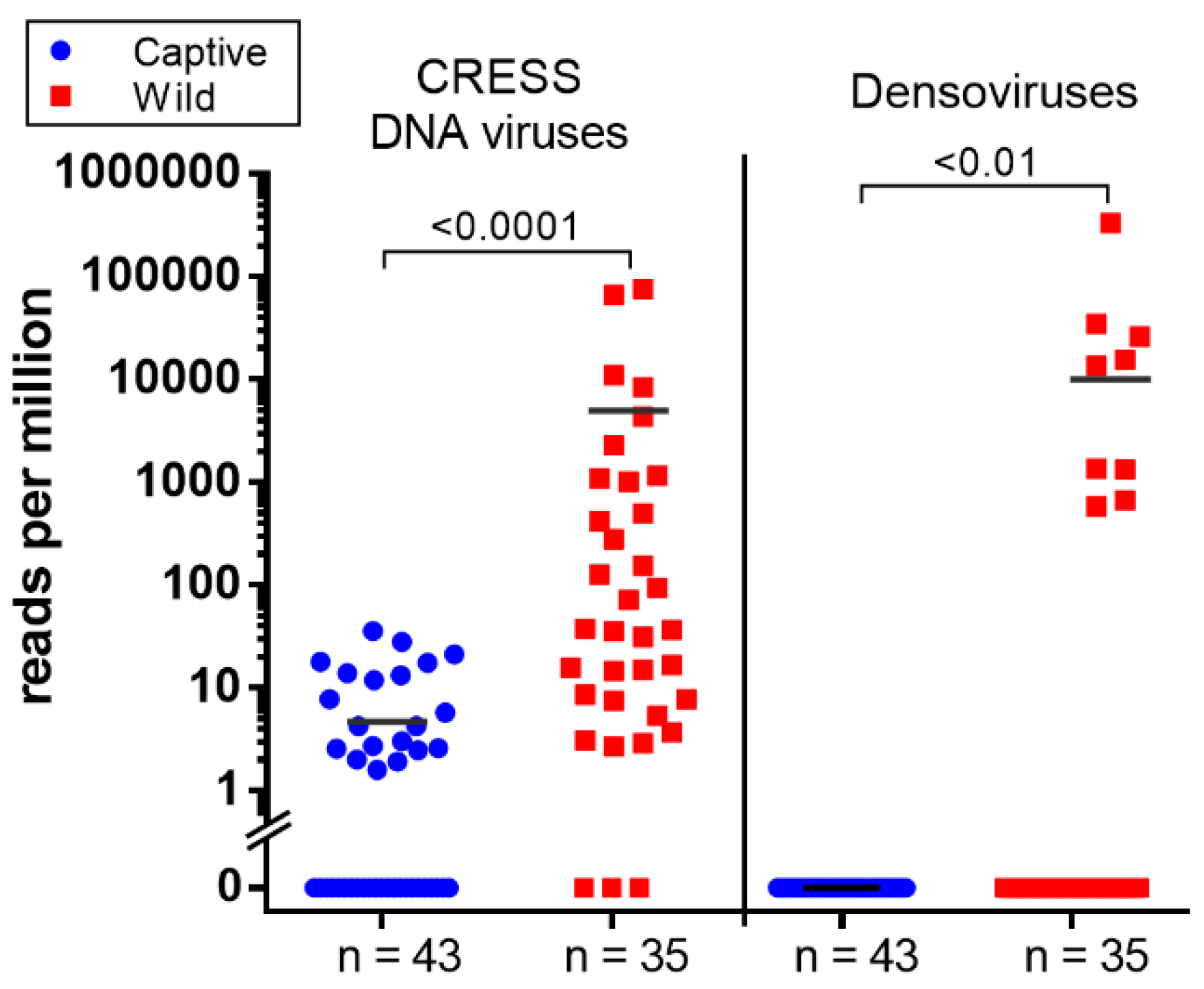
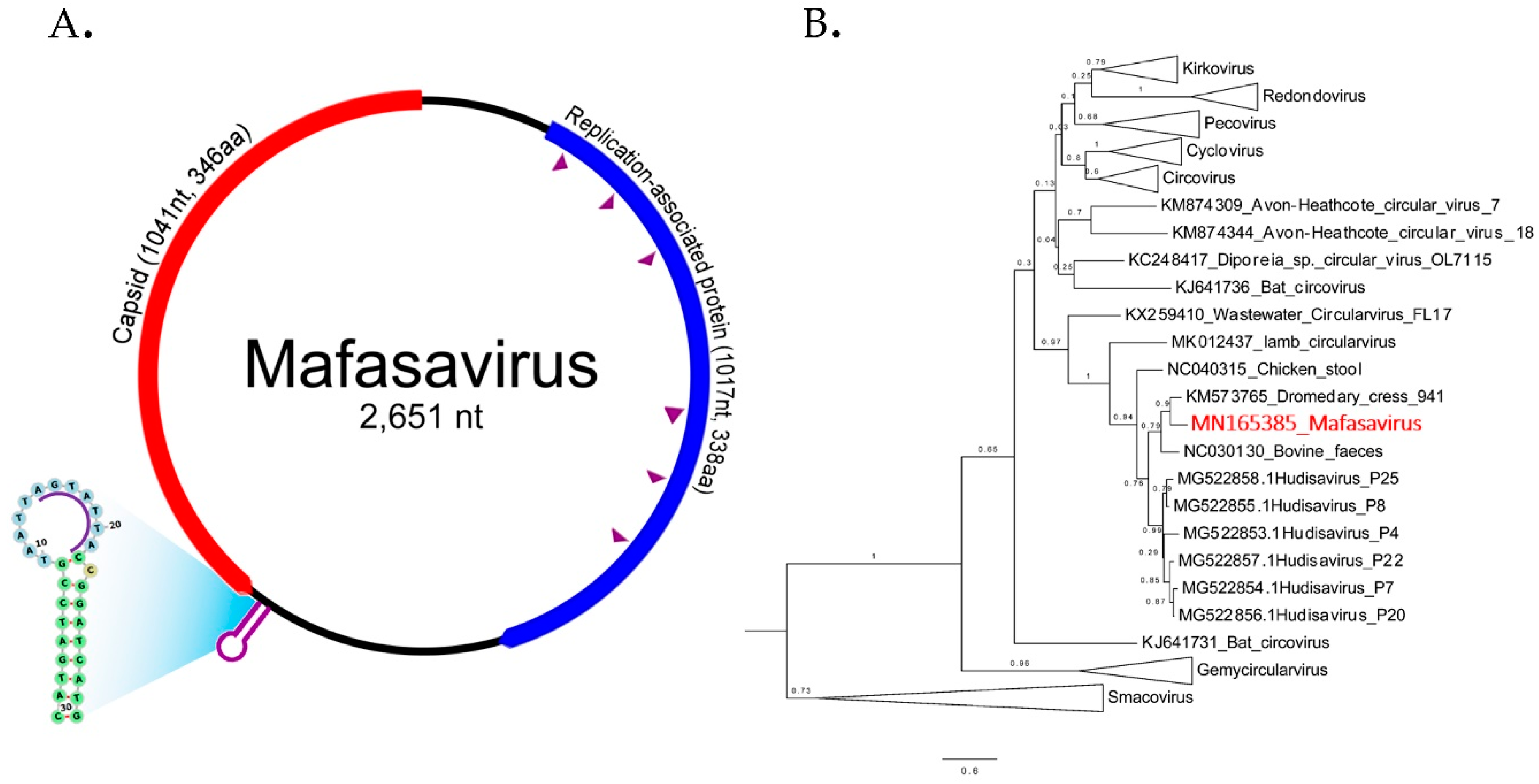
3.6. CRESS-DNA Virus
Novel Stool-Associated CRESS DNA Virus
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carding, S.R.; Davis, N.; Hoyles, L. Review article: The human intestinal virome in health and disease. Aliment. Pharmacol. Ther. 2017, 46, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Virgin, H.W. The virome in mammalian physiology and disease. Cell 2014, 157, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Neil, J.A.; Cadwell, K. The Intestinal Virome and Immunity. J. Immunol. 2018, 201, 1615–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhya, I.; Segal, J.P.; Carding, S.R.; Hart, A.L.; Hold, G.L. The gut virome: The ‘missing link’ between gut bacteria and host immunity? Therap. Adv. Gastroenterol. 2019, 12, 1756284819836620. [Google Scholar] [CrossRef]
- Santiago-Rodriguez, T.M.; Hollister, E.B. Human Virome and Disease: High-Throughput Sequencing for Virus Discovery, Identification of Phage-Bacteria Dysbiosis and Development of Therapeutic Approaches with Emphasis on the Human Gut. Viruses 2019, 11, 656. [Google Scholar] [CrossRef]
- Delwart, E. Animal virus discovery: Improving animal health, understanding zoonoses, and opportunities for vaccine development. Curr. Opin. Virol. 2012, 2, 344–352. [Google Scholar] [CrossRef]
- Li, L.; Shan, T.; Wang, C.; Cote, C.; Kolman, J.; Onions, D.; Gulland, F.M.; Delwart, E. The fecal viral flora of California sea lions. J. Virol. 2011, 85, 9909–9917. [Google Scholar] [CrossRef]
- Handley, S.A.; Thackray, L.B.; Zhao, G.; Presti, R.; Miller, A.D.; Droit, L.; Abbink, P.; Maxfield, L.F.; Kambal, A.; Duan, E.; et al. Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell 2012, 151, 253–266. [Google Scholar] [CrossRef]
- Zhang, W.; Li, L.; Deng, X.; Kapusinszky, B.; Pesavento, P.A.; Delwart, E. Faecal virome of cats in an animal shelter. J. Gen. Virol. 2014, 95, 2553–2564. [Google Scholar] [CrossRef]
- Phan, T.G.; Kapusinszky, B.; Wang, C.; Rose, R.K.; Lipton, H.L.; Delwart, E.L. The fecal viral flora of wild rodents. PLoS Pathog. 2011, 7, e1002218. [Google Scholar] [CrossRef]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Rabiela, F.; Wiratsudakul, A.; Suzan, G.; Rico-Chavez, O. Viral networks and detection of potential zoonotic viruses in bats and rodents: A worldwide analysis. Zoonoses Public Health 2019, 66, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Biek, R.; Walsh, P.D.; Leroy, E.M.; Real, L.A. Recent common ancestry of Ebola Zaire virus found in a bat reservoir. PLoS Pathog. 2006, 2, e90. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Hahn, B.H. The evolution of HIV-1 and the origin of AIDS. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 2487–2494. [Google Scholar] [CrossRef]
- Han, H.J.; Yu, H.; Yu, X.J. Evidence for zoonotic origins of Middle East respiratory syndrome coronavirus. J. Gen. Virol. 2016, 97, 274–280. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Fan, R.Y.Y.; Luk, H.K.H.; Zhu, L.; Fung, J.; Li, K.S.M.; Wong, E.Y.M.; Ahmed, S.S.; Chan, J.F.W.; Kok, R.K.H.; et al. Replication of MERS and SARS coronaviruses in bat cells offers insights to their ancestral origins. Emerg. Microbes Infect. 2018, 7, 209. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Wong, A.C.P.; Lau, T.C.K.; Woo, P.C.Y. Molecular Evolution of MERS Coronavirus: Dromedaries as a Recent Intermediate Host or Long-Time Animal Reservoir? Int. J. Mol. Sci. 2017, 18, 2138. [Google Scholar] [CrossRef]
- Mohd, H.A.; Al-Tawfiq, J.A.; Memish, Z.A. Middle East Respiratory Syndrome Coronavirus (MERS-CoV) origin and animal reservoir. Virol. J. 2016, 13, 87. [Google Scholar] [CrossRef]
- De Graaf, M.; Beck, R.; Caccio, S.M.; Duim, B.; Fraaij, P.L.; Le Guyader, F.S.; Lecuit, M.; Le Pendu, J.; De Wit, E.; Schultsz, C. Sustained fecal-oral human-to-human transmission following a zoonotic event. Curr. Opin. Virol. 2017, 22, 1–6. [Google Scholar] [CrossRef]
- Fong, T.-T.; Lipp, E.K. Enteric viruses of humans and animals in aquatic environments: Health risks, detection, and potential water quality assessment tools. Microbiol. Mol. Biol. Rev. 2005, 69, 357–371. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Lu, X.J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019, 68, 1169–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Fooden, J. Systematic review of Southeast Asia longtail macaques, Macaca fascicularis (Raffles,1821). Fieldiana Zool NS 1995, 81, 1–206. [Google Scholar]
- Kyes, R.C. Survey of the long-tailed macaques introduced onto Tinjil Island, Indonesia. Am. J. Primatol. 1993, 31, 77–83. [Google Scholar] [CrossRef]
- Itoh, Y. Translational research on influenza virus infection using a nonhuman primate model. Pathol. Int. 2016, 66, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.; Mendoza, E.J.; Plummer, F.A.; Gao, G.F.; Kobinger, G.P.; Qiu, X. From bench to almost bedside: The long road to a licensed Ebola virus vaccine. Expert Opin. Biol. Ther. 2018, 18, 159–173. [Google Scholar] [CrossRef]
- Antony, J.M.; MacDonald, K.S. A critical analysis of the cynomolgus macaque, Macaca fascicularis, as a model to test HIV-1/SIV vaccine efficacy. Vaccine 2015, 33, 3073–3083. [Google Scholar] [CrossRef]
- Bailey, C.; Mansfield, K. Emerging and reemerging infectious diseases of nonhuman primates in the laboratory setting. Vet. Pathol. 2010, 47, 462–481. [Google Scholar] [CrossRef]
- Rosshart, S.P.; Herz, J.; Vassallo, B.G.; Hunter, A.; Wall, M.K.; Badger, J.H.; McCulloch, J.A.; Anastasakis, D.G.; Sarshad, A.A.; Leonardi, I.; et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 2019, 365, eaaw4361. [Google Scholar] [CrossRef]
- Holly Smith, B.; Crummett, T.L.; Brandt, K.L. Ages of eruption of primate teeth: A compendium for aging individuals and comparing life histories. Am. J. Phys. Anthropol. 1994, 37, 177–231. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Deng, X.; Mee, E.T.; Collot-Teixeira, S.; Anderson, R.; Schepelmann, S.; Minor, P.D.; Delwart, E. Comparing viral metagenomics methods using a highly multiplexed human viral pathogens reagent. J. Virol. methods 2015, 213, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; McGinnis, S.; Madden, T.L. BLAST: Improvements for better sequence analysis. Nucleic Acids Res. 2006, 34, W6–W9. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Naccache, S.N.; Ng, T.; Federman, S.; Li, L.; Chiu, C.Y.; Delwart, E.L. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res. 2015, 43, e46. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Hales, L.M.; Knowles, N.J.; Reddy, P.S.; Xu, L.; Hay, C.; Hallenbeck, P.L. Complete genome sequence analysis of Seneca Valley virus-001, a novel oncolytic picornavirus. J. Gen. Virol. 2008, 89, 1265–1275. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Kerpedjiev, P.; Hammer, S.; Hofacker, I.L. Forna (force-directed RNA): Simple and effective online RNA secondary structure diagrams. Bioinformatics 2015, 31, 3377–3379. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, T.R.; Dhote, V.; Yu, Y.; Hellen, C.U. A distinct class of internal ribosomal entry site in members of the Kobuvirus and proposed Salivirus and Paraturdivirus genera of the Picornaviridae. J. Virol. 2012, 86, 1468–1486. [Google Scholar] [CrossRef]
- Pestova, T.V.; Shatsky, I.N.; Hellen, C.U. Functional dissection of eukaryotic initiation factor 4F: The 4A subunit and the central domain of the 4G subunit are sufficient to mediate internal entry of 43S preinitiation complexes. Mol. Cell Biol. 1996, 16, 6870–6878. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ling, Y.; Shan, T.; Yang, S.; Xu, H.; Deng, X.; Delwart, E.; Zhang, W. Gut virome of mammals and birds reveals high genetic diversity of the family Microviridae. Virus Evol. 2019, 5, vez013. [Google Scholar] [CrossRef] [PubMed]
- Boros, A.; Polgar, B.; Pankovics, P.; Fenyvesi, H.; Engelmann, P.; Phan, T.G.; Delwart, E.; Reuter, G. Multiple divergent picobirnaviruses with functional prokaryotic Shine-Dalgarno ribosome binding sites present in cloacal sample of a diarrheic chicken. Virology 2018, 525, 62–72. [Google Scholar] [CrossRef]
- Krishnamurthy, S.R.; Wang, D. Extensive conservation of prokaryotic ribosomal binding sites in known and novel picobirnaviruses. Virology 2018, 516, 108–114. [Google Scholar] [CrossRef]
- Staheli, J.P.; Dyen, M.R.; Deutsch, G.H.; Basom, R.S.; Fitzgibbon, M.P.; Lewis, P.; Barcy, S. Complete Unique Genome Sequence, Expression Profile, and Salivary Gland Tissue Tropism of the Herpesvirus 7 Homolog in Pigtailed Macaques. J. Virol. 2016, 90, 6657–6674. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.; Dang, J.; Bzhalava, D.; Stern, J.; Edelstein, Z.R.; Koutsky, L.A.; Kiviat, N.B.; Feng, Q. Characterization of three novel human papillomavirus types isolated from oral rinse samples of healthy individuals. J. Clin. Virol. 2014, 59, 30–37. [Google Scholar] [CrossRef]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Roediger, B.; Lee, Q.; Tikoo, S.; Cobbin, J.C.A.; Henderson, J.M.; Jormakka, M.; O’Rourke, M.B.; Padula, M.P.; Pinello, N.; Henry, M.; et al. An Atypical Parvovirus Drives Chronic Tubulointerstitial Nephropathy and Kidney Fibrosis. Cell 2018, 175, 530–543. [Google Scholar] [CrossRef]
- Penzes, J.J.; de Souza, W.M.; Agbandje-McKenna, M.; Gifford, R.J. An Ancient Lineage of Highly Divergent Parvoviruses Infects both Vertebrate and Invertebrate Hosts. Viruses 2019, 11, 525. [Google Scholar] [CrossRef] [PubMed]
- Kapusinszky, B.; Ardeshir, A.; Mulvaney, U.; Deng, X.; Delwart, E. Case-Control Comparison of Enteric Viromes in Captive Rhesus Macaques with Acute or Idiopathic Chronic Diarrhea. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altan, E.; Del Valle Mendoza, J.; Deng, X.; Phan, T.G.; Sadeghi, M.; Delwart, E.L. Small Circular Rep-Encoding Single-Stranded DNA Genomes in Peruvian Diarrhea Virome. Genome Announc 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Hsu, B.B.; Gibson, T.E.; Yeliseyev, V.; Liu, Q.; Lyon, L.; Bry, L.; Silver, P.A.; Gerber, G.K. Dynamic Modulation of the Gut Microbiota and Metabolome by Bacteriophages in a Mouse Model. Cell Host Microbe 2019, 25, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Aggarwala, V.; Liang, G.; Bushman, F.D. Viral communities of the human gut: Metagenomic analysis of composition and dynamics. Mob. DNA 2017, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Blanton, L.V.; Cao, S.; Zhao, G.; Manary, M.; Trehan, I.; Smith, M.I.; Wang, D.; Virgin, H.W.; Rohwer, F.; et al. Gut DNA viromes of Malawian twins discordant for severe acute malnutrition. Proc. Natl. Acad. Sci. USA 2015, 112, 11941–11946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat guano virome: Predominance of dietary viruses from insects and plants plus novel mammalian viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar] [CrossRef]
- Fernandes, M.A.; Verstraete, S.G.; Phan, T.G.; Deng, X.; Stekol, E.; LaMere, B.; Lynch, S.V.; Heyman, M.B.; Delwart, E. Enteric Virome and Bacterial Microbiota in Children with Ulcerative Colitis and Crohn Disease. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 30–36. [Google Scholar] [CrossRef]
- Krupovic, M.; Forterre, P. Microviridae goes temperate: Microvirus-related proviruses reside in the genomes of Bacteroidetes. PLoS ONE 2011, 6, e19893. [Google Scholar] [CrossRef]
- Manrique, P.; Bolduc, B.; Walk, S.T.; van der Oost, J.; de Vos, W.M.; Young, M.J. Healthy human gut phageome. Proc. Natl. Acad. Sci. USA 2016, 113, 10400–10405. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Krupovic, M.; Poulet, A.; Debroas, D.; Enault, F. Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS ONE 2012, 7, e40418. [Google Scholar] [CrossRef] [PubMed]
- Arvin, A.; Campadelli-Fiume, G.; Mocarski, E.; Moore, P.S.; Roizman, B.; Whitley, R.; Yamanishi, K. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Blatter, J.A.; Sweet, S.C.; Conrad, C.; Danziger-Isakov, L.A.; Faro, A.; Goldfarb, S.B.; Hayes, D., Jr.; Melicoff, E.; Schecter, M.; Storch, G.; et al. Anellovirus loads are associated with outcomes in pediatric lung transplantation. Pediatric Transplant. 2018, 22. [Google Scholar] [CrossRef] [PubMed]
- De Vlaminck, I.; Khush, K.K.; Strehl, C.; Kohli, B.; Luikart, H.; Neff, N.F.; Okamoto, J.; Snyder, T.M.; Cornfield, D.N.; Nicolls, M.R.; et al. Temporal response of the human virome to immunosuppression and antiviral therapy. Cell 2013, 155, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Deng, X.; Linsuwanon, P.; Bangsberg, D.; Bwana, M.B.; Hunt, P.; Martin, J.N.; Deeks, S.G.; Delwart, E. AIDS alters the commensal plasma virome. J. Virol. 2013, 87, 10912–10915. [Google Scholar] [CrossRef]
- Young, J.C.; Chehoud, C.; Bittinger, K.; Bailey, A.; Diamond, J.M.; Cantu, E.; Haas, A.R.; Abbas, A.; Frye, L.; Christie, J.D.; et al. Viral metagenomics reveal blooms of anelloviruses in the respiratory tract of lung transplant recipients. Am. J. Transplant. 2015, 15, 200–209. [Google Scholar] [CrossRef]
- Bauer, E.; Williams, B.A.; Smidt, H.; Verstegen, M.W.; Mosenthin, R. Influence of the gastrointestinal microbiota on development of the immune system in young animals. Curr. Issues Intest. Microbiol. 2006, 7, 35–51. [Google Scholar]
- Elmore, D.; Eberle, R. Monkey B virus (Cercopithecine herpesvirus 1). Comp. Med. 2008, 58, 11–21. [Google Scholar]
- Dhurandhar, N.V.; Dhurandhar, E.J.; Ingram, D.K.; Vaughan, K.; Mattison, J.A. Natural infection of human adenovirus 36 in rhesus monkeys is associated with a reduction in fasting glucose 36. J. Diabetes 2014, 6, 614–616. [Google Scholar] [CrossRef]
- Sukmak, M.; Wajjwalku, W.; Ostner, J.; Schulke, O. A first report of non-invasive adenovirus detection in wild Assamese macaques in Thailand. Primates 2017, 58, 307–313. [Google Scholar] [CrossRef]
- Roy, S.; Sandhu, A.; Medina, A.; Clawson, D.S.; Wilson, J.M. Adenoviruses in fecal samples from asymptomatic rhesus macaques, United States. Emerg. Infect. Dis. 2012, 18, 1081–1088. [Google Scholar] [CrossRef]
- Wevers, D.; Metzger, S.; Babweteera, F.; Bieberbach, M.; Boesch, C.; Cameron, K.; Couacy-Hymann, E.; Cranfield, M.; Gray, M.; Harris, L.A.; et al. Novel adenoviruses in wild primates: A high level of genetic diversity and evidence of zoonotic transmissions. J. Virol. 2011, 85, 10774–10784. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Yagi, S.; Carrion, R., Jr.; Chen, E.C.; Liu, M.; Brasky, K.M.; Lanford, R.E.; Kelly, K.R.; Bales, K.L.; Schnurr, D.P.; et al. Experimental cross-species infection of common marmosets by titi monkey adenovirus. PLoS ONE 2013, 8, e68558. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.C.; Yagi, S.; Kelly, K.R.; Mendoza, S.P.; Tarara, R.P.; Canfield, D.R.; Maninger, N.; Rosenthal, A.; Spinner, A.; Bales, K.L.; et al. Cross-species transmission of a novel adenovirus associated with a fulminant pneumonia outbreak in a new world monkey colony. PLoS Pathog. 2011, 7, e1002155. [Google Scholar] [CrossRef]
- Palinski, R.M.; Mitra, N.; Hause, B.M. Discovery of a novel Parvovirinae virus, porcine parvovirus 7, by metagenomic sequencing of porcine rectal swabs. Virus Genes 2016, 52, 564–567. [Google Scholar] [CrossRef]
- Streck, A.F.; Canal, C.W.; Truyen, U. Molecular epidemiology and evolution of porcine parvoviruses. Infect. Genet. Evol. 2015, 36, 300–306. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, K.K.; Wang, J.; Wang, X.P.; Zhao, L.; Sun, P.; Li, Y.D. Detection and molecular characterization of novel porcine parvovirus 7 in Anhui province from Central-Eastern China. Infect. Genet. Evol. 2019, 71, 31–35. [Google Scholar] [CrossRef]
- Ouh, I.O.; Park, S.; Lee, J.Y.; Song, J.Y.; Cho, I.S.; Kim, H.R.; Park, C.K. First detection and genetic characterization of porcine parvovirus 7 from Korean domestic pig farms. J. Vet. Sci. 2018, 19, 855–857. [Google Scholar] [CrossRef]
- Xiao, C.T.; Halbur, P.G.; Opriessnig, T. Complete Genome Sequence of a Novel Porcine Parvovirus (PPV) Provisionally Designated PPV5. Genome Announc. 2013, 1. [Google Scholar] [CrossRef]
- Blinkova, O.; Victoria, J.; Li, Y.; Keele, B.F.; Sanz, C.; Ndjango, J.B.; Peeters, M.; Travis, D.; Lonsdorf, E.V.; Wilson, M.L.; et al. Novel circular DNA viruses in stool samples of wild-living chimpanzees. J. Gen. Virol. 2010, 91, 74–86. [Google Scholar] [CrossRef]
- Varsani, A.; Krupovic, M. Smacoviridae: A new family of animal-associated single-stranded DNA viruses. Arch. Virol. 2018, 163, 2005–2015. [Google Scholar] [CrossRef]
- Li, L.; McGraw, S.; Zhu, K.; Leutenegger, C.M.; Marks, S.L.; Kubiski, S.; Gaffney, P.; Dela Cruz, F.N., Jr.; Wang, C.; Delwart, E.; et al. Circovirus in tissues of dogs with vasculitis and hemorrhage. Emerg. Infect. Dis. 2013, 19, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Liu, Y.; Li, N.; Wang, Y.; Zhang, S.; Hu, R. Novel circovirus from mink, China. Emerg. Infect. Dis. 2014, 20, 1548–1550. [Google Scholar] [CrossRef] [PubMed]
- Segales, J.; Kekarainen, T.; Cortey, M. The natural history of porcine circovirus type 2: From an inoffensive virus to a devastating swine disease? Vet. Microbiol. 2013, 165, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, J.D.; Dominguez-Bello, M.G.; Contreras, M.; Lander, O.; Caballero-Arias, H.; Xutao, D.; Noya-Alarcon, O.; Delwart, E. Complex virome in feces from Amerindian children in isolated Amazonian villages. Nat. Commun. 2018, 9, 4270. [Google Scholar] [CrossRef] [PubMed]
- Kassim, N.; Hambali, K.; Amir, A. Nutritional Composition of Fruits Selected by Long-Tailed Macaques (Macaca fascicularis) in Kuala Selangor, Malaysia. Trop. Life Sci. Res. 2017, 28, 91–101. [Google Scholar] [CrossRef]
- Handley, S.A.; Desai, C.; Zhao, G.; Droit, L.; Monaco, C.L.; Schroeder, A.C.; Nkolola, J.P.; Norman, M.E.; Miller, A.D.; Wang, D.; et al. SIV Infection-Mediated Changes in Gastrointestinal Bacterial Microbiome and Virome Are Associated with Immunodeficiency and Prevented by Vaccination. Cell Host Microbe 2016, 19, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Droit, L.; Gilbert, M.H.; Schiro, F.R.; Didier, P.J.; Si, X.; Paredes, A.; Handley, S.A.; Virgin, H.W.; Bohm, R.P.; et al. Virome biogeography in the lower gastrointestinal tract of rhesus macaques with chronic diarrhea. Virology 2019, 527, 77–88. [Google Scholar] [CrossRef]
- Anthony, S.J.; Islam, A.; Johnson, C.; Navarrete-Macias, I.; Liang, E.; Jain, K.; Hitchens, P.L.; Che, X.; Soloyvov, A.; Hicks, A.L.; et al. Non-random patterns in viral diversity. Nat. Commun. 2015, 6, 8147. [Google Scholar] [CrossRef] [Green Version]
- Riesland, N.J.; Wilde, H. Expert Review of Evidence Bases for Managing Monkey Bites in Travelers. J. Travel Med. 2015, 22, 259–262. [Google Scholar] [CrossRef] [Green Version]
- Eberle, R.; Jones-Engel, L. Understanding Primate Herpesviruses. J. Emerg. Dis. Virol. 2017, 3. [Google Scholar] [CrossRef]
- Cohen, J.I.; Davenport, D.S.; Stewart, J.A.; Deitchman, S.; Hilliard, J.K.; Chapman, L.E.; Group, B.V.W. Recommendations for prevention of and therapy for exposure to B virus (cercopithecine herpesvirus 1). Clin. Infect. Dis. 2002, 35, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- Johnston, W.F.; Yeh, J.; Nierenberg, R.; Procopio, G. Exposure to Macaque Monkey Bite. J. Emerg. Med. 2015, 49, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Blaise, A.; Parola, P.; Brouqui, P.; Gautret, P. Rabies Postexposure Prophylaxis for Travelers Injured by Nonhuman Primates, Marseille, France, 2001–2014. Emerg. Infect. Dis. 2015, 21, 1473–1476. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | All | Herpes B Virus-Positive (+) | Herpes B Virus-Negative (−) | p-Values (Herpes B Positive vs Negative) | p-Values (Captive vs Wild) | ||
|---|---|---|---|---|---|---|---|
| Age (years) a | captive | 3.34 ± 1.65 | 4.68 ± 1.40 | 2.27 ± 0.88 | <0.001 *** | <0.05 * | |
| wild c | 5.48 ± 4.11 | 6.14 ± 4.27 | 2.75 ± 1.31 | <0.01 ** | |||
| Sex (%) b | captive | M | 20 (46.5%) | 9 (20.9%) | 11 (25.6%) | 0.74 | 0.24 |
| F | 23 (53.5%) | 10 (23.3%) | 13 (30.2%) | ||||
| wild c | M | 19 (61.29%) | 16 (51.61%) | 3 (9.68%) | 0.53 | ||
| F | 12 (38.71%) | 9 (29.03%) | 3 (9.68%) | ||||
| Bodyweight a | captive | 3.13 ± 1.21 | 3.95 ± 1.04 | 2.49 ± 0.91 | <0.001 *** | <0.05 * | |
| wild c | 4.16 ± 1.90 | 4.05 ± 1.90 | 2.78 ± 1.11 | <0.05 * | |||
| Crown–Rump Length a | captive | 386.6 ± 58.4 | 428.4 ± 45.8 | 353.5 ± 44.4 | <0.001 *** | <0.05 * | |
| wild c | 415.4 ± 55.5 | 425.5 ± 51.9 | 371.5 ± 47.8 | 0.053 | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sawaswong, V.; Fahsbender, E.; Altan, E.; Kemthong, T.; Deng, X.; Malaivijitnond, S.; Payungporn, S.; Delwart, E. High Diversity and Novel Enteric Viruses in Fecal Viromes of Healthy Wild and Captive Thai Cynomolgus Macaques (Macaca fascicularis). Viruses 2019, 11, 971. https://doi.org/10.3390/v11100971
Sawaswong V, Fahsbender E, Altan E, Kemthong T, Deng X, Malaivijitnond S, Payungporn S, Delwart E. High Diversity and Novel Enteric Viruses in Fecal Viromes of Healthy Wild and Captive Thai Cynomolgus Macaques (Macaca fascicularis). Viruses. 2019; 11(10):971. https://doi.org/10.3390/v11100971
Chicago/Turabian StyleSawaswong, Vorthon, Elizabeth Fahsbender, Eda Altan, Taratorn Kemthong, Xutao Deng, Suchinda Malaivijitnond, Sunchai Payungporn, and Eric Delwart. 2019. "High Diversity and Novel Enteric Viruses in Fecal Viromes of Healthy Wild and Captive Thai Cynomolgus Macaques (Macaca fascicularis)" Viruses 11, no. 10: 971. https://doi.org/10.3390/v11100971
APA StyleSawaswong, V., Fahsbender, E., Altan, E., Kemthong, T., Deng, X., Malaivijitnond, S., Payungporn, S., & Delwart, E. (2019). High Diversity and Novel Enteric Viruses in Fecal Viromes of Healthy Wild and Captive Thai Cynomolgus Macaques (Macaca fascicularis). Viruses, 11(10), 971. https://doi.org/10.3390/v11100971