Identification of Broad-Spectrum Antiviral Compounds by Targeting Viral Entry

 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses, Virus-Like Particles (VLPs), and Infection
2.2. Cells
2.3. Compounds
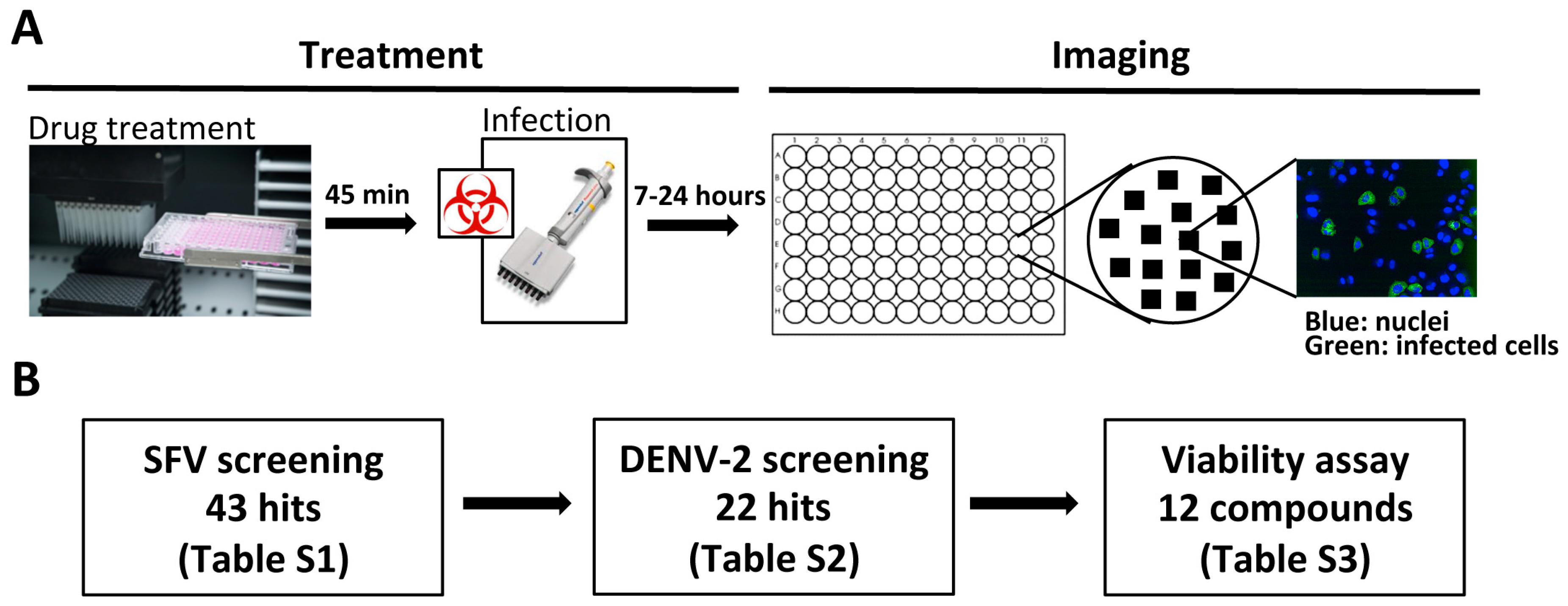
2.4. Phenotypic Screening
2.5. Immunofluorescence Staining
2.6. MTT Assay
2.7. IC50 and TC50 Calculation
2.8. β-Lactamase Assay
2.9. Semliki Forest Virus (SFV) Cell Binding Assay
2.10. SFV Internalisation Assay
2.11. SFV Envelope Acidification Assay
2.12. SFV Lipid Mixing Assay
2.13. Exogenous Protein Expression Measurement
2.14. Endogenous RNA Transcription Measurement
2.15. Transferrin Internalisation
2.16. In Vivo Pharmacokinetics
2.17. Mice and Infections
2.18. Zika Virus (ZIKV) RNA Quantification
2.19. Immunofocus Assay
2.20. Staining of ZIKV Infected Cells and Flow Cytometry Analysis
2.21. Safety/Biosecurity
2.22. Statement on Animal Ethics
3. Results
3.1. Identification of Broad-Spectrum Antiviral Compounds
3.2. Selected Compounds Display Broad-Spectrum Activity against Other Alpha- and Flavi-Viruses
3.3. Selected Compounds Display Broad-Spectrum Activity against Viruses from Different Viral Families
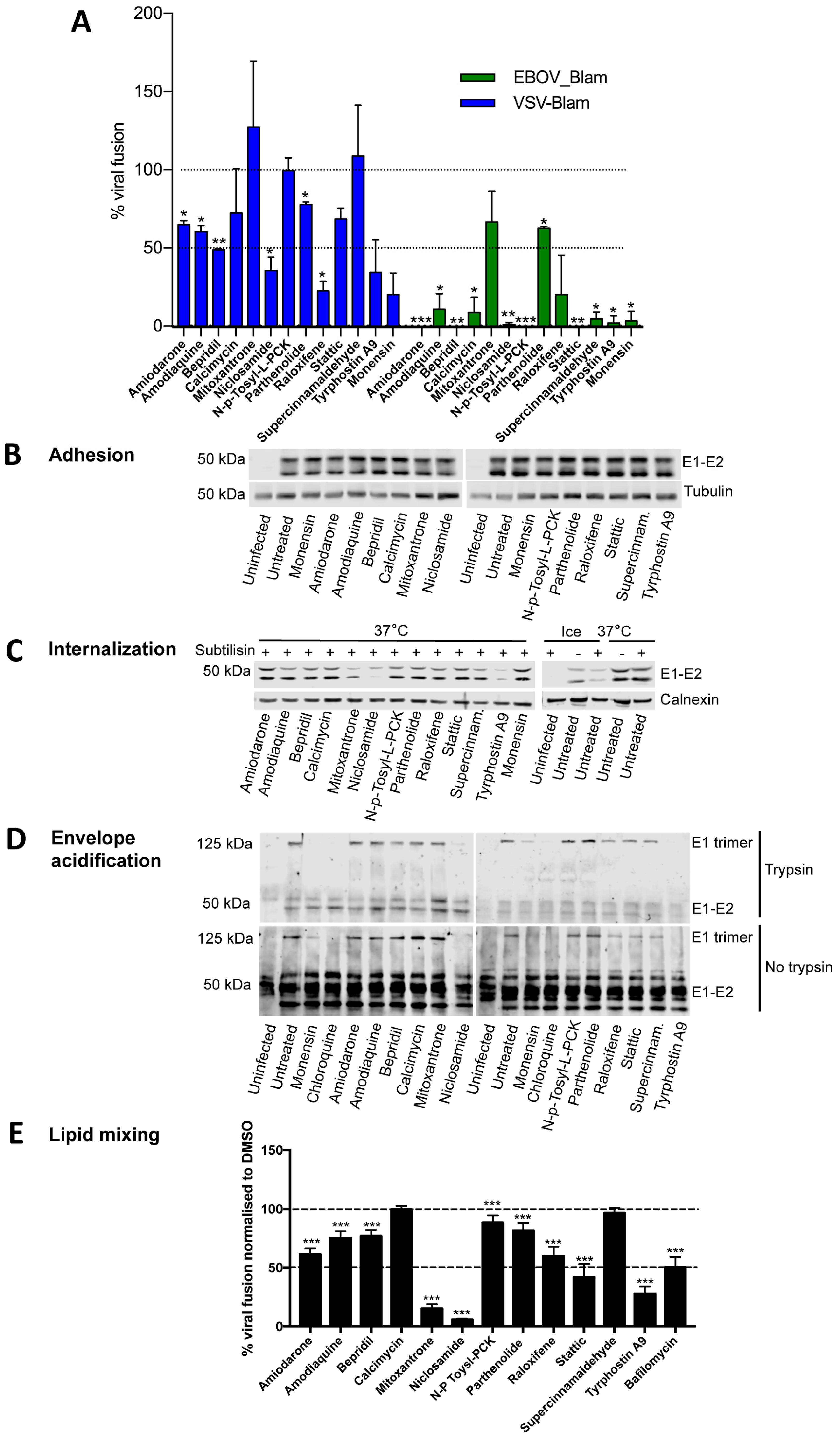
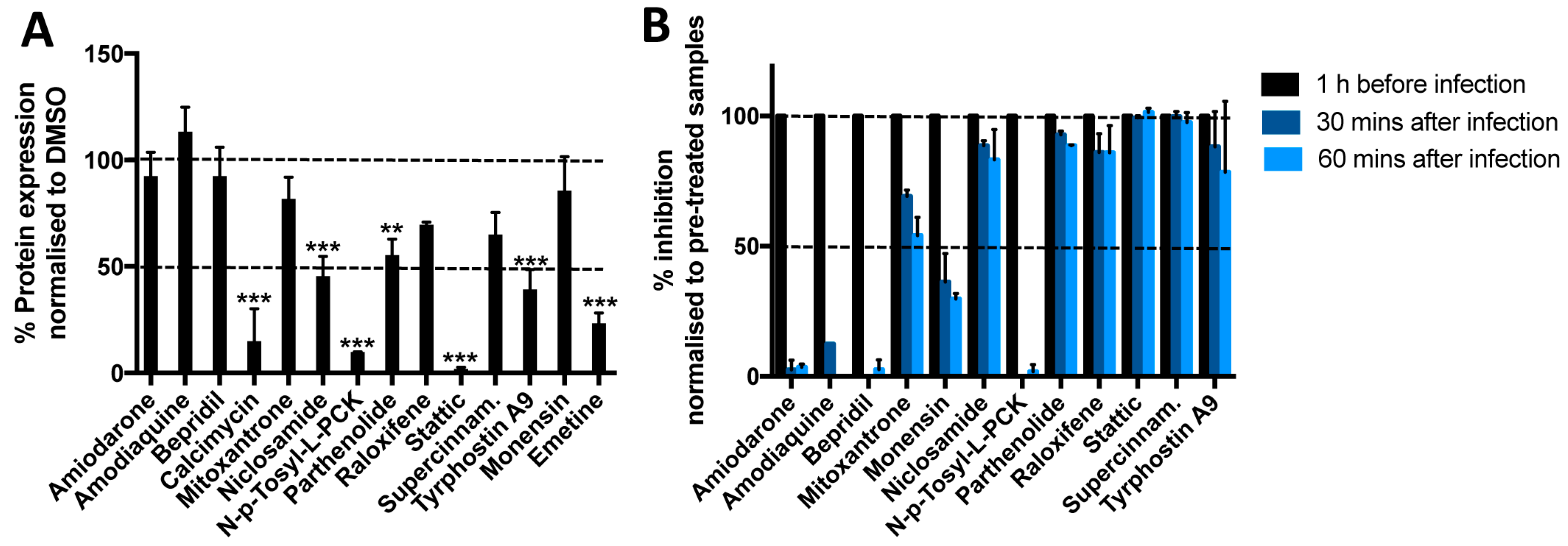
3.4. Most Compounds Inhibit Infection before or at the Stage of Fusion
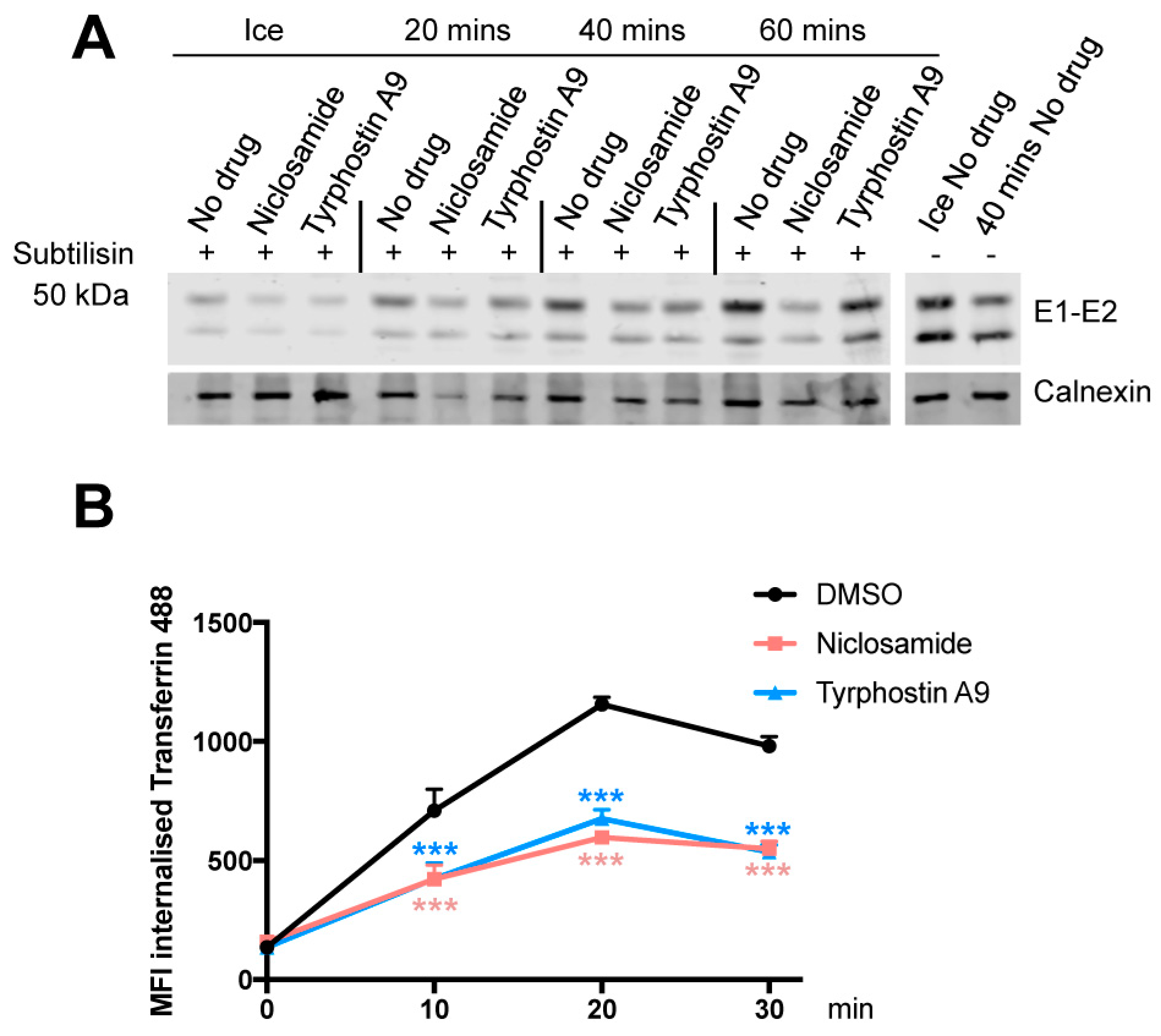
3.5. Niclosamide and Tyrphostin A9 Inhibit Viral Entry by Reducing Clathrin-Mediated Endocytosis
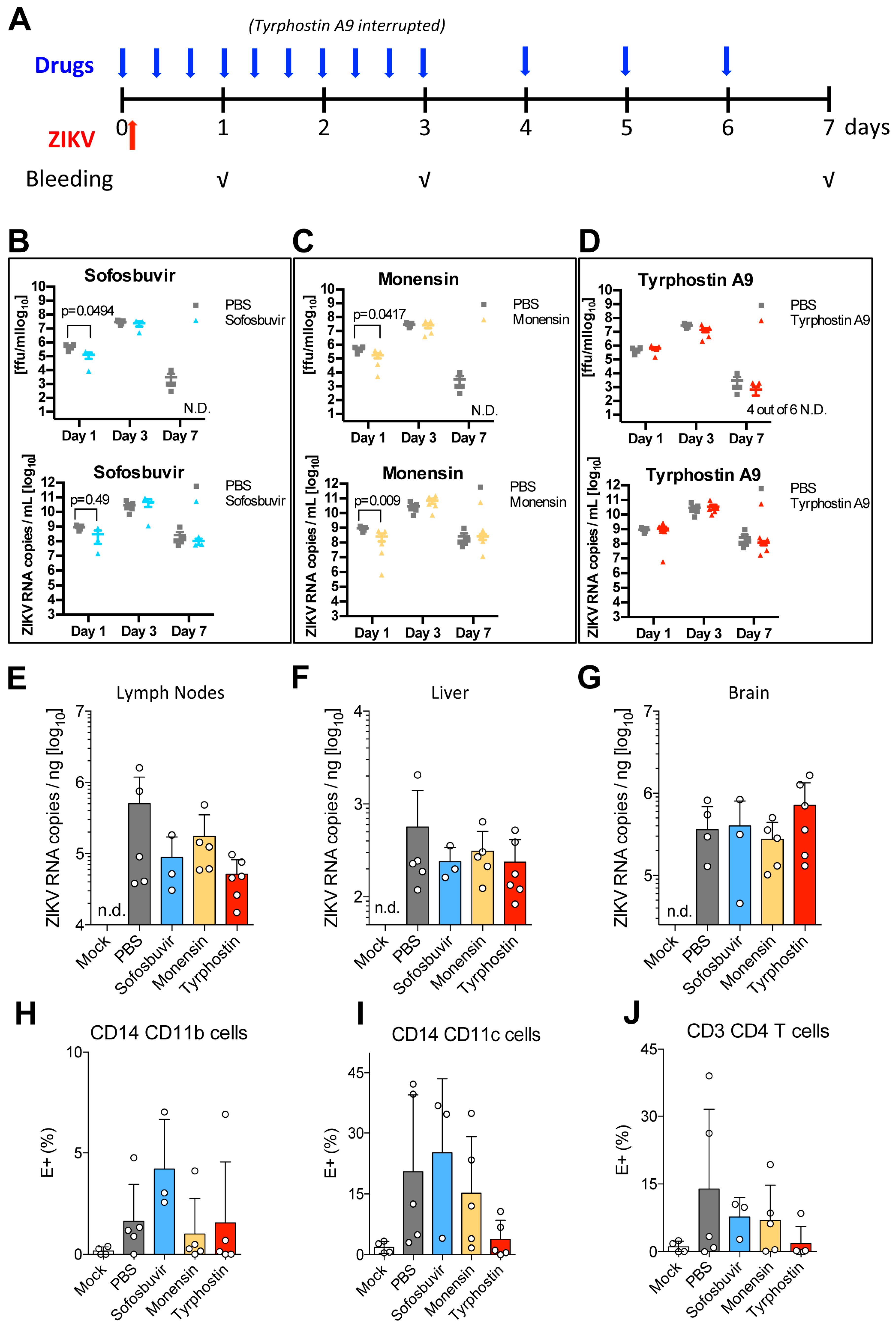
3.6. Tyrphostin A9 and Monensin Reduce Viral Titres in the Blood and in the Lymph Nodes
4. Discussion
5. Conclusions
Dedication
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Coltart, C.E.M.; Lindsey, B.; Ghinai, I.; Johnson, A.M.; Heymann, D.L. The ebola outbreak, 2013–2016: Old lessons for new epidemics. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Barcellos, C.; Brasil, P.; Cruz, O.G.; Honório, N.A.; Kuper, H.; Carvalho, M.S. The zika virus epidemic in brazil: From discovery to future implications. Int. J. Environ. Res. Public Health 2018, 15, 96. [Google Scholar] [CrossRef] [PubMed]
- Paules, C.I.; Fauci, A.S. Yellow fever—Once again on the radar screen in the Americas. N. Engl. J. Med. 2017, 376, 1397–1399. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, T.; Mamahit, A.; Cox, N.J. 65 years of influenza surveillance by a world health organization-coordinated global network. Influenza Other Respi. Viruses 2018, 12, 558–565. [Google Scholar] [CrossRef]
- Wahid, B.; Ali, A.; Rafique, S.; Idrees, M. Global expansion of chikungunya virus: Mapping the 64-year history. Int. J. Infect. Dis. 2017, 58, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Bekerman, E.; Einav, S. Combating emerging viral threats. Science 2015, 348, 282–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.D.; Meng, W.; Wang, X.J.; Wang, H.C. Broad-spectrum antiviral agents. Front. Microbiol. 2015, 6, 517. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.E.; Dorhoi, A.; Hotchkiss, R.S.; Bartenschlager, R. Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discov. 2017, 17, 35. [Google Scholar] [CrossRef]
- Zhou, Y.; Simmons, G. Development of novel entry inhibitors targeting emerging viruses. Expert Rev. Anti Infect. Ther. 2012, 10, 1129–1138. [Google Scholar] [CrossRef] [Green Version]
- Marsh, M.; Helenius, A. Virus entry: Open sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.F.; Kolokoltsov, A.A.; Albrecht, T.; Davey, R.A. Cellular entry of ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog. 2010, 6, e1001110. [Google Scholar] [CrossRef] [PubMed]
- Markosyan, R.M.; Miao, C.; Zheng, Y.-M.; Melikyan, G.B.; Liu, S.-L.; Cohen, F.S. Induction of cell-cell fusion by ebola virus glycoprotein: Low ph is not a trigger. PLoS Pathog. 2016, 12, e1005373. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, G.; Horvath, P.; Schweingruber, C.; Zünd, D.; McInerney, G.; Merits, A.; Mühlemann, O.; Azzalin, C.; Helenius, A. The host nonsense-mediated mrna decay pathway restricts mammalian rna virus replication. Cell Host Microbe 2014, 16, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, M.; Castro, C.; Thaa, B.; Liu, L.; Mutso, M.; Liu, X.; Mahalingam, S.; Griffin, J.L.; Marsh, M.; McInerney, G.M. Alphavirus-induced hyperactivation of pi3k/akt directs pro-viral metabolic changes. PLoS Pathog. 2018, 14, e1006835. [Google Scholar] [CrossRef] [PubMed]
- Giese, S.; Marsh, M. Tetherin can restrict cell-free and cell-cell transmission of hiv from primary macrophages to t cells. PLoS Pathog. 2014, 10, e1004189. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, R.; Nagy, D.; Mandel, R.J.; Naldini, L.; Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 1997, 15, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.; McNabb, S.; Goddard, T.; Horton, D.L.; Lembo, T.; Nel, L.H.; Weiss, R.A.; Cleaveland, S.; Fooks, A.R. A robust lentiviral pseudotype neutralisation assay for in-field serosurveillance of rabies and lyssaviruses in africa. Vaccine 2009, 27, 7178–7186. [Google Scholar] [CrossRef]
- Mallajosyula, V.V.A.; Citron, M.; Ferrara, F.; Lu, X.; Callahan, C.; Heidecker, G.J.; Sarma, S.P.; Flynn, J.A.; Temperton, N.J.; Liang, X.; et al. Influenza hemagglutinin stem-fragment immunogen elicits broadly neutralizing antibodies and confers heterologous protection. Proc. Natl. Acad. Sci. USA 2014, 111, E2514–E2523. [Google Scholar] [CrossRef]
- Niwa, H.; Yamamura, K.; Miyazaki, J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 1991, 108, 193–199. [Google Scholar]
- Tscherne, D.M.; Manicassamy, B.; Garcia-Sastre, A. An enzymatic virus-like particle assay for sensitive detection of virus entry. J. Virol. Methods 2010, 163, 336–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, M.; Bron, R. Sfv infection in cho cells: Cell-type specific restrictions to productive virus entry at the cell surface. J. Cell Sci. 1997, 110 (Pt 1), 95–103. [Google Scholar] [PubMed]
- Unterholzner, L.; Sumner, R.P.; Baran, M.; Ren, H.; Mansur, D.S.; Bourke, N.M.; Randow, F.; Smith, G.L.; Bowie, A.G. Vaccinia virus protein c6 is a virulence factor that binds tbk-1 adaptor proteins and inhibits activation of irf3 and irf7. PLoS Pathog. 2011, 7, e1002247. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.; Yip, C.C.; Tee, K.M.; Zhu, Z.; Tsang, J.O.; Chik, K.K.; Tsang, T.G.; Chan, C.C.; Poon, V.K.; Sridhar, S.; et al. Improved detection of zika virus rna in human and animal specimens by a novel, highly sensitive and specific real-time rt-pcr assay targeting the 5′-untranslated region of zika virus. Trop. Med. Int. Health 2017, 22, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Marsh, M.; Wellsteed, J.; Kern, H.; Harms, E.; Helenius, A. Monensin inhibits semliki forest virus penetration into culture cells. Proc. Natl. Acad. Sci. USA 1982, 79, 5297–5301. [Google Scholar] [CrossRef] [PubMed]
- Martín, C.S.-S.; Liu, C.Y.; Kielian, M. Dealing with low ph: Entry and exit of alphaviruses and flaviviruses. Trends Microbiol. 2009, 17, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Albertini, A.A.V.; Baquero, E.; Ferlin, A.; Gaudin, Y. Molecular and cellular aspects of rhabdovirus entry. Viruses 2012, 4, 117–139. [Google Scholar] [CrossRef] [PubMed]
- Luo, M. Influenza virus entry. In Viral Molecular Machines; Rossmann, M.G., Rao, V.B., Eds.; Springer: Boston, MA, USA, 2012; pp. 201–221. [Google Scholar]
- Stein, B.S.; Gowda, S.D.; Lifson, J.D.; Penhallow, R.C.; Bensch, K.G.; Engleman, E.G. PH-independent HIV entry into CD4-positive T cells via virus envelope fusion to the plasma membrane. Cell 1987, 49, 659–668. [Google Scholar] [CrossRef]
- McClure, M.O.; Marsh, M.; Weiss, R.A. Human immunodeficiency virus infection of cd4-bearing cells occurs by a ph-independent mechanism. EMBO J. 1988, 7, 513–518. [Google Scholar] [CrossRef]
- Koyama, A.H.; Uchida, T. The mode of entry of herpes simplex virus type 1 into vero cells. Microbiol. Immunol. 1987, 31, 123–130. [Google Scholar] [CrossRef]
- Mercer, J.; Helenius, A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 2008, 320, 531. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.S.; Buley, T.; Evison, B.J.; Cutts, S.M.; Neumann, G.M.; Iskander, M.N.; Phillips, D.R. A molecular understanding of mitoxantrone-DNA adduct formation: Effect of cytosine methylation and flanking sequences. J. Biol. Chem. 2004, 279, 18814–18823. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, E.N.; Sigova, A.A.; Zinchenko, V.P. Mechanism of action of calcium ionophores on intact cells: Ionophore-resistant cells. Membr. Cell Biol. 2000, 13, 357–368. [Google Scholar] [PubMed]
- Helenius, A.; Kartenbeck, J.; Simons, K.; Fries, E. On the entry of semliki forest virus into bhk-21 cells. J. Cell Biol. 1980, 84, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Kielian, M.; Helenius, A. Ph-induced alterations in the fusogenic spike protein of semliki forest virus. J. Cell Biol. 1985, 101, 2284. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Nunez, N.V.; Wilschut, J.; Smit, J.M. Monitoring virus entry into living cells using did-labeled dengue virus particles. Methods 2011, 55, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Lakadamyali, M.; Rust, M.J.; Babcock, H.P.; Zhuang, X. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. USA 2003, 100, 9280–9285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, S.R.; Hernandez, R.; Brown, D.T. Role of the vacuolar-atpase in sindbis virus infection. J. Virol. 2011, 85, 1257–1266. [Google Scholar] [CrossRef]
- Ye, Y.; Zhang, X.; Zhang, T.; Wang, H.; Wu, B. Design and evaluation of injectable niclosamide nanocrystals prepared by wet media milling technique. Drug Dev. Ind. Pharm. 2015, 41, 1416–1424. [Google Scholar] [CrossRef]
- Classes of pesticides. In Handbook of Pesticide Toxicology; Hayes, W.J., Jr.; Laws, E.R., Jr. (Eds.) Academic press: New York, NY, USA, 1991; Volume 3, p. 1497. [Google Scholar]
- Aliota, M.T.; Caine, E.A.; Walker, E.C.; Larkin, K.E.; Camacho, E.; Osorio, J.E. Characterization of lethal zika virus infection in ag129 mice. PLoS Negl. Trop. Dis. 2016, 10, e0004682. [Google Scholar] [CrossRef]
- Zellweger, R.M.; Shresta, S. Mouse models to study dengue virus immunology and pathogenesis. Front. Immunol. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- EMEA. Committee for Medicinal Products for Veterinary Use. Available online: https://www.ema.europa.eu/documents/mrl-report/monensin-cattle-including-dairy-cows-summary-report-committee-veterinary-medicinal-products_en.pdf (accessed on 18 February 2019).
- Kirby, B.J.; Symonds, W.T.; Kearney, B.P.; Mathias, A.A. Pharmacokinetic, pharmacodynamic, and drug-interaction profile of the hepatitis C virus NS5b polymerase inhibitor sofosbuvir. Clin. Pharmacokinet. 2015, 54, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Sacramento, C.Q.; de Melo, G.R.; de Freitas, C.S.; Rocha, N.; Hoelz, L.V.B.; Miranda, M.; Fintelman-Rodrigues, N.; Marttorelli, A.; Ferreira, A.C.; Barbosa-Lima, G.; et al. The clinically approved antiviral drug sofosbuvir inhibits zika virus replication. Sci. Rep. 2017, 7, 40920. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.C.; Zaverucha-do-Valle, C.; Reis, P.A.; Barbosa-Lima, G.; Vieira, Y.R.; Mattos, M.; Silva, P.d.P.; Sacramento, C.; de Castro Faria Neto, H.C.; Campanati, L.; et al. Sofosbuvir protects zika virus-infected mice from mortality, preventing short- and long-term sequelae. Sci. Rep. 2017, 7, 9409. [Google Scholar] [CrossRef] [PubMed]
- Johansen, L.M.; DeWald, L.E.; Shoemaker, C.J.; Hoffstrom, B.G.; Lear-Rooney, C.M.; Stossel, A.; Nelson, E.; Delos, S.E.; Simmons, J.A.; Grenier, J.M.; et al. A screen of approved drugs and molecular probes identifies therapeutics with anti–ebola virus activity. Sci. Transl. Med. 2015, 7, ra289–ra290. [Google Scholar] [CrossRef] [PubMed]
- Salata, C.; Baritussio, A.; Munegato, D.; Calistri, A.; Ha, H.R.; Bigler, L.; Fabris, F.; Parolin, C.; Palu, G.; Mirazimi, A. Amiodarone and metabolite mdea inhibit ebola virus infection by interfering with the viral entry process. Pathog. Dis. 2015, 73. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Mesplede, T.; Xu, H.; Quan, Y.; Wainberg, M.A. The antimalarial drug amodiaquine possesses anti-zika virus activities. J. Med. Virol. 2018, 90, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Turone, F. Doctors trial amiodarone for ebola in sierra leone. BMJ 2014, 349. [Google Scholar] [CrossRef] [PubMed]
- Cross, R.W.; Mire, C.E.; Feldmann, H.; Geisbert, T.W. Post-exposure treatments for Ebola and Marburg virus infections. Nat. Rev. Drug Discov. 2018, 17, 413. [Google Scholar] [CrossRef] [PubMed]
- Banbury, D.N.; Oakley, J.D.; Sessions, R.B.; Banting, G. Tyrphostin a23 inhibits internalization of the transferrin receptor by perturbing the interaction between tyrosine motifs and the medium chain subunit of the ap-2 adaptor complex. J. Biol. Chem. 2003, 278, 12022–12028. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Brecher, M.; Deng, Y.-Q.; Zhang, J.; Sakamuru, S.; Liu, B.; Huang, R.; Koetzner, C.A.; Allen, C.A.; Jones, S.A.; et al. Existing drugs as broad-spectrum and potent inhibitors for zika virus by targeting ns2b-ns3 interaction. Cell Res. 2017, 27, 1046–1064. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Lee, E.M.; Wen, Z.; Cheng, Y.; Huang, W.-K.; Qian, X.; Tcw, J.; Kouznetsova, J.; Ogden, S.C.; Hammack, C.; et al. Identification of small-molecule inhibitors of zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016, 22, 1101. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.-C.; HuangFu, W.-C.; Tsai, T.-T.; Ho, M.-R.; Jhan, M.-K.; Shen, T.-J.; Tseng, P.-C.; Wang, Y.-T.; Lin, C.-F. The antiparasitic drug niclosamide inhibits dengue virus infection by interfering with endosomal acidification independent of mtor. PLoS Negl. Trop. Dis. 2018, 12, e0006715. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-M.; Lu, J.-W.; Lin, C.-C.; Chin, Y.-F.; Wu, T.-Y.; Lin, L.-I.; Lai, Z.-Z.; Kuo, S.-C.; Ho, Y.-J. Antiviral activities of niclosamide and nitazoxanide against chikungunya virus entry and transmission. Antiviral Res. 2016, 135, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Jurgeit, A.; McDowell, R.; Moese, S.; Meldrum, E.; Schwendener, R.; Greber, U.F. Niclosamide is a proton carrier and targets acidic endosomes with broad antiviral effects. PLoS Pathog. 2012, 8, e1002976. [Google Scholar] [CrossRef] [PubMed]
- Dejonghe, W.; Kuenen, S.; Mylle, E.; Vasileva, M.; Keech, O.; Viotti, C.; Swerts, J.; Fendrych, M.; Ortiz-Morea, F.A.; Mishev, K.; et al. Mitochondrial uncouplers inhibit clathrin-mediated endocytosis largely through cytoplasmic acidification. Nat. Commun. 2016, 7, 11710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edinger, T.O.; Pohl, M.O.; Stertz, S. Entry of influenza a virus: Host factors and antiviral targets. J. Gen. Virol. 2014, 95, 263–277. [Google Scholar] [CrossRef] [PubMed]
- De La Vega, M.-A.; Caleo, G.; Audet, J.; Qiu, X.; Kozak, R.A.; Brooks, J.I.; Kern, S.; Wolz, A.; Sprecher, A.; Greig, J.; et al. Ebola viral load at diagnosis associates with patient outcome and outbreak evolution. J. Clin. Investig. 2015, 125, 4421–4428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughn, D.W.; Green, S.; Kalayanarooj, S.; Innis, B.L.; Nimmannitya, S.; Suntayakorn, S.; Endy, T.P.; Raengsakulrach, B.; Rothman, A.L.; Ennis, F.A.; et al. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J. Infect. Dis. 2000, 181, 2–9. [Google Scholar] [CrossRef]
- Thi, E.P.; Lee, A.C.H.; Geisbert, J.B.; Ursic-Bedoya, R.; Agans, K.N.; Robbins, M.; Deer, D.J.; Fenton, K.A.; Kondratowicz, A.S.; MacLachlan, I.; et al. Rescue of non-human primates from advanced sudan ebolavirus infection with lipid encapsulated siRNA. Nat. Microbiol. 2016, 1, 16142. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SFV (7 h) (µM) | DENV-2 (24 h) (µM) | DENV-2 (7 + 17 h ) (µM) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 | TC50 | SI | IC50 | TC50 | SI | IC50 | TC50 | SI | |
| Amiodarone | 9 | 96.8 | 10.8 | 12.7 | ~25.5 | 2.0 | 15.5 | 40.6 | 2.6 |
| Amodiaquine | 8.5 | >50 | >5.8 | 17.2 | 18.4 | 1.1 | 12.7 | 44.2 | 3.5 |
| Bepridil | 9.5 | 34.9 | 3.7 | 12.1 | ~23.4 | 1.9 | 15.16 | ~48.8 | 3.2 |
| Calcimycin | 0.6 | 4.1 | 6.8 | 0.38 | 6.7 | 17.6 | 4.4 | >50 | >11.3 |
| Mitoxantrone | 7.0 | 43.2 | 6.2 | 2.1 | 4.6 | 2.2 | 2.0 | 7.9 | 4.0 |
| Monensin (cntrl) | 1.1 | >50 | >45 | 3.8 | >50 | 13.2 | 4.5 | >50 | >11.1 |
| Niclosamide | 3 | >50 | >16.7 | 0.4 | 12.8 | 32.0 | 1.1 | >50 | >45.5 |
| N-p-Tosyl-L-PKC | 10.7 | 42.7 | 4.0 | 7.5 | 31.7 | 4.2 | 19.6 | 24.2 | 1.2 |
| Parthenolide | 3.3 | 11.7 | 3.5 | 7.5 | 11.9 | 1.6 | 12.2 | 35.8 | 2.9 |
| Raloxifene | 15.1 | >50 | >3.3 | 9.8 | ~22.63 | 2.3 | 5.7 | 24.8 | 4.4 |
| Stattic | 3.7 | 7.8 | 2.1 | 0.9 | 9.7 | 10.8 | 3 | 17.3 | 5.8 |
| Supercinnamaldehyde | 15.8 | 40.5 | 2.6 | 15.0 | 25.8 | 1.7 | 15.4 | 59.6 | 3.9 |
| Tyrphostin A9 | 2.9 | >50 | >17.2 | 0.4 | 28.5 | 71.2 | 1.2 | >50 | >41.7 |
| SINV (µM) | RRV (µM) | DENV-1 (µM) | DENV-3 (µM) | ZIKV (µM) | YF17D (µM) | HCV (µM) | |
|---|---|---|---|---|---|---|---|
| Amiodarone | ~11.8 | 3.2 | ~20 | ~25 | No inhibition | 8.12 | 3.9 |
| Amodiaquine | 11.3 | 6.2 | ~24.8 | ~25 | No inhibition | 5.3 | 4.2 |
| Bepridil | 14.6 | 4.6 | ~20 | ~20 | ~25.2 | 7.1 | ~12.4 |
| Calcimycin | 2.1 | 6.5 | 4.3 | NA | 17.1 | 0.4 | 1.45 |
| Mitoxantrone | 9.9 | 4.9 | 5.6 | 6.3 | ~25 | 2.2 | 2.7 |
| Monensin (cntrl) | 0.4 | 4.6 | 6.2 | ~3.6 | 5.5 | < 0.3 | 2.5 |
| Niclosamide | 2.8 | 0.82 | 1.5 | 1.6 | ~0.7 | < 0.3 | 0.2 |
| N-p-Tosyl-L-PCK | 11.9 | ~6.3 | 25.6 | 24.2 | 24.3 | 4.6 | ~13.0 |
| Parthenolide | 3.2 | 7.7 | 10 | 15.8 | 6.1 | 6.4 | 5.6 |
| Raloxifene | 30.6 | ND | 8.8 | 5.3 | ND | 7.8 | 3.9 |
| Stattic | 3.6 | 3.8 | 3 | NA | 4 | 4.9 | 3.7 |
| Supercinnamaldehyde | 24.9 | 18.9 | ~22.7 | 12.2 | No inhibition | 7.6 | 15.8 |
| Tyrphostin A9 | ~0.8 | 0.6 | 1.4 | 1.2 | <0.3 | < 0.3 | <0.3 |
| VSV | RABV | H5N1 | EBOV | HIV | HSV (Vero) | VACV | |
|---|---|---|---|---|---|---|---|
| System | VLP | VLP | VLP | VLP | virus | virus | virus |
| Family | Rhabdov. | Rhabdov. | Orthomyxov. | Filov. | Retrov. | Herpesv. | Poxv. |
| Time of assay | 24 h | 24 h | 48 h | 48 h | 24 h | 8 h | 8 h |
| Entry mechanism | CME | CME | CME | Macrop. | PM fusion (?) | PM fusion | Macrop. |
| Fusion | EE | EE | LE | LE | PM (?) | PM | EE |
| low pH dependency | Yes | Yes | Yes | Yes | No | No | No |
| Amiodarone | 25 | No inhibition | 12.5 | 0.9 | No inhibition | 24.6 | No inhibition |
| Amodiaquine | 11.2 | 23.5 | 13.6 | 0.9 | No inhibition | 28.22 | No inhibition |
| Bepridil | 10.4 | 6.9 | 8.2 | 0.8 | No inhibition | No inhibition | 26.8 |
| Calcimycin | 0.1 | 0.7 | 0.4 | 0.5 | 2.5 | <0.5 | NA |
| Mitoxantrone | 0.6 | <0.5 | 2.8 | 0.1 | NA | 6.8 | No inhibition |
| Monensin (cntrl) | 0.7 | 0.8 | 0.9 | 4.5 | No inhibition | No inhibition | No inhibition |
| Niclosamide | 3.5 | 1.3 | 1.4 | 2.9 | 3.3 | 1.3 | 3 |
| N-p-Tosyl-L-PCK | 11.5 | 15 | No inhibition | No inhibition | 12.5 | No inhibition | 14.4 |
| Parthenolide | 2.6 | 3.8 | 19.4 | Toxic | 3.7 | No inhibition | 5 |
| Raloxifene | 12.7 | No inhibition | No inhibition | 1.4 | No inhibition | 23.5 | 20 |
| Stattic | 3.0 | 3.2 | Toxic | Toxic | 3.2 | 5.2 | 4.1 |
| Supercinnam. | 14.6 | 21.6 | 17.5 | 21.2 | No inhibition | 25.2 | No inhibition |
| Tyrphostin A9 | 4.1 | 1.4 | <0.5 | 3.2 | 1.1 | 1.5 | 3.4 |
| Adhesion | Internal. | E1 Acid. | Translation | Time of Addition | Hemifusion | Fusion | Viruses Inhibited | ||
|---|---|---|---|---|---|---|---|---|---|
| VSV_Blam | EBOV_Blam | ||||||||
| Amiodarone | X | X | X | X | Before fusion | V | V | V | Endosomal pH-dep. |
| Amodiaquine | X | X | X | X | Before fusion | V | V | V | Endosomal pH-dep. |
| Bepridil | X | X | X | X | Before fusion | V | V | V | Endosomal pH-dep. |
| Calcimycin | X | X | X | V | After fusion | X | X | V | All tested |
| Mitoxantrone | X | X | X | X | Before and after fusion | ND | X | X | Unclear pattern |
| Niclosamide | X | V | V | V | Before and after fusion | V | V | V | All tested |
| N-p-Tosyl-L-PCK | X | X | X | V | Before fusion | X | X | V | Unclear pattern |
| Parthenolide | X | X | X | V | Before and after fusion | X | X | V | Unclear pattern |
| Raloxifene | X | X | X | X | Before and after fusion | V | V | V | Unclear pattern |
| Stattic | X | X | X | V | After fusion | V | X | V | All tested |
| Supercinnamaldehyde | X | X | X | X | After fusion | X | X | V | Unclar pattern |
| Tyrphostin A9 | X | V | V | V | Before and after fusion | V | V | V | All tested |
| Compound | Suggested Mechanisms of Action |
|---|---|
| Amiodarone | Inhibition of fusion (lipid mixing) |
| Amodiaquine | Inhibition of fusion (lipid mixing) |
| Bepridil | Inhibition of fusion (lipid mixing) |
| Calcimycin | Inhibition of translation and unknown mechanisms |
| Mitoxantrone | Unknown mechanisms |
| Niclosamide | Inhibitor of viral internalisation and translation |
| N-p-Tosyl-L-PCK | Inhibitor of proteases important for different viruses |
| Parthenolide | Inhibition of translation and unknown mechanisms |
| Raloxifene | Inhibition of fusion (lipid mixing) |
| Stattic | Inhibition of lipid mixing, translation and unknown mechanisms |
| Supercinnamaldehyde | Unknown mechanisms |
| Tyrphostin A9 | Inhibitor of viral internalisation and translation |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzon, M.; Ortega-Prieto, A.M.; Imrie, D.; Luft, C.; Hess, L.; Czieso, S.; Grove, J.; Skelton, J.K.; Farleigh, L.; Bugert, J.J.; et al. Identification of Broad-Spectrum Antiviral Compounds by Targeting Viral Entry. Viruses 2019, 11, 176. https://doi.org/10.3390/v11020176
Mazzon M, Ortega-Prieto AM, Imrie D, Luft C, Hess L, Czieso S, Grove J, Skelton JK, Farleigh L, Bugert JJ, et al. Identification of Broad-Spectrum Antiviral Compounds by Targeting Viral Entry. Viruses. 2019; 11(2):176. https://doi.org/10.3390/v11020176
Chicago/Turabian StyleMazzon, Michela, Ana Maria Ortega-Prieto, Douglas Imrie, Christin Luft, Lena Hess, Stephanie Czieso, Joe Grove, Jessica Katy Skelton, Laura Farleigh, Joachim J. Bugert, and et al. 2019. "Identification of Broad-Spectrum Antiviral Compounds by Targeting Viral Entry" Viruses 11, no. 2: 176. https://doi.org/10.3390/v11020176
APA StyleMazzon, M., Ortega-Prieto, A. M., Imrie, D., Luft, C., Hess, L., Czieso, S., Grove, J., Skelton, J. K., Farleigh, L., Bugert, J. J., Wright, E., Temperton, N., Angell, R., Oxenford, S., Jacobs, M., Ketteler, R., Dorner, M., & Marsh, M. (2019). Identification of Broad-Spectrum Antiviral Compounds by Targeting Viral Entry. Viruses, 11(2), 176. https://doi.org/10.3390/v11020176