1. Introduction
Crimean-Congo hemorrhagic fever virus (CCHFV), a member of the
Orthonairovirus genus and
Nairoviridae family, causes a hemorrhagic fever disease with considerable mortality rates in endemic areas such as Turkey [
1,
2]. The widespread distribution, significant mortality rates and lack of specific treatment or control measures make CCHFV an urgent target for vaccine development [
3,
4].
The CCHFV nucleocapsid (N) protein is an important structural component with the potential to stimulate humoral and cellular responses following natural or experimental infections. It has, therefore, been targeted for vaccine development using DNA and viral vector platforms in several studies [
5,
6,
7]. During challenge experiments in different modified mice models, such as IFNα/β/γR−/−, immune-suppressed (IS) and STAT-1 knock-out mice, vaccinations based on this protein have demonstrated protective effects in lethal challenges despite the lack of neutralizing antibodies [
6,
8]. Different expression platforms such as adenovirus and modified vaccinia Ankara (MVA)-based vectors were previously used to deliver N proteins with inconclusive results. A recombinant DNA-based CCHFV-N expression system was previously developed, using modified vaccinia Ankara vectors as the delivery system. However, the immune responses in vaccinated mice were not evaluated in detail and lacked cytokine and antibody response analyses. Although this system seemed to induce antigen-specific immunogenicity in mice, it failed to exert any protective effects upon virus challenge [
7].
Considering the disadvantages of routinely used expression platforms, such as pre-existing immunity, the possibility of genome integration, immune responses to the antigens of viral vectors and toxicity related to lytic infections [
9], the exploration of new platforms for N protein delivery is a credible approach and likely to accelerate CCHFV vaccine development efforts.
Bovine herpesvirus type 4 (BoHV-4), a member of the
gammaherpesvirinae subfamily and
rhadinovirus genus, is prevalent among the cattle population, and can be isolated from healthy animals as well as those with diverse clinical symptoms, although there is no clear evidence linking this virus to any specific pathology [
10,
11,
12]. While the potential of BoHV-4 to infect humans in vivo is obscure, its oncolytic potential based on selective replication strategies in some human cancer cell lines has been demonstrated in vitro [
13,
14]. This virus has been successfully explored as a novel herpesviral vector in targeting different viral immunodominant antigens, such as the Ebola surface glycoprotein (BoHV-4-syEBOVgD106ΔTK), Peste des Petits Ruminants virus hemagglutinin (H) (BoHV-4-A-PPRV-H-ΔTK) and various glycoproteins (A29L, M1R and B6R) of Monkeypox virus (BoHV-4-A-CMV-A29LgD
106ΔTK, BoHV-4-A-EF1α-M1RgD
106ΔTK and BoHV-4-A-EF1α-B6RgD
106ΔTK) due to some interesting characteristics. These include easy propagation in cell culture systems, a relatively simple genome with a large capacity for inserting foreign genes, availability of an animal model for investigating its pathogenicity (rabbit) and a lack of documented cell transformation in comparison to other
gammaherpesviruses [
15,
16,
17,
18,
19,
20].
In this study, we generated a recombinant Bovine herpesvirus type 4, expressing the N protein of CCHFV (BoHV4-∆TK-CCHFV-N), and evaluated its immunogenicity in BALB/c and protection potential through a lethal challenge experiment in IFNα/β/γR−/− mice models. Due to the mentioned advantages of this viral vector, we assumed that this novel expression platform could be considered as an alternative in CCHFV vaccination. To test our hypothesis, we further compared the efficacy of BoHV4-∆TK-CCHFV-N with two widely used delivery platforms, including Adenovirus type 5 (Ad5-N) alongside a DNA vector (pCD-N1) expressing the same antigen.
2. Methods
2.1. Ethics Statement
All animal experiments were performed with the official permission of the Ankara University Ethical Committee for Animal Experiments (17/12/2014; 2014-23-155 and 17/10/2018; 2018-20-130). All animal samplings were conducted according to the national regulations on the operation and procedure of animal experiment ethics committees (Regulation Nr.26220, Date:09.7.2006). During these studies, humane endpoint scores were considered. Multiple observations per day were conducted to confirm the animals’ welfare. Constant access to sterilized water and food was provided. Mice were euthanized by CO2 exposure and cervical dislocation. BALB/c and Interferon alpha/beta/gamma receptor-null mice (IFNα/β/γR−/−; AG129; Strain: 129S7/SvEvBrd) were provided by B&K Universal Ltd. (Marshall Bioresouces, Hull, East Yorkshire, UK).
2.2. Viruses and Cells
A bovine herpesvirus type 4 (BoHV-4)-carrying bacterial artificial chromosome vector (BoHV4-BAC) was developed from the Movar33/63 European strain by the insertion of a F plasmid (pBeloBAC11; NEB, MA, USA) containing a bacterial and mammalian selection cassette (loxp-BAC-EGFP-loxp) between the open reading frame 2 (ORF2) and ORF3 genes (GenBank Accession Number: NC002665), which are not essential for virus replication, by in vitro homologous recombination in Madin–Darby bovine kidney (MDBK) cells and the infectious BAC (iBAC or BoHV4-BAC). The CCHFV Ank-2 strain (GenBank Accession Number: MK309333; third passage in SW-13 cells) was used as the challenge virus. CCHFV and BoHV4-BAC were cultivated in Scott and White No. 13 (SW-13) and MDBK cells, respectively. SW-13 and MDBK cells were cultured in Eagle’s Minimum Essential Medium (EMEM; Sigma, St. Louis, MO, USA) and Leibovitz’s L-15 (Thermo Fisher Scientific, Waltham, MA, USA) media, supplemented with 10% fetal bovine serum (FBS; Biological Industries, Kibbutz Beit-Haemek, Israel), 2 mM L-glutamine (Biological Industries, Kibbutz Beit-Haemek, Israel), 100 U penicillin, and 0.1 mg/mL streptomycin (Thermo Fisher Scientific). Baby hamster kidney (BHK21-C13) and MDBK stably expressing Cre recombinase enzyme (MDBK-cre) cells were used in the transfection assays and loxp-BAC-EGFP-loxp elimination from the BoHV4-BAC construct, respectively. Cultivation of these cell lines was the same as for MDBK cells. In addition, we performed preliminary experiments using human embryonic kidney (HEK293 and HEK293A) cells for the generation and cultivation of the recombinant adenovirus type 5 expressing CCHFV N protein. These cells were cultured and maintained at 37 °C/5% CO2 in Minimum Essential Medium alpha (Thermo Fisher Scientific). The final N-expressing constructs, including BoHV4-∆TK-CCHFV-N and Ad5-N, were propagated and titered in MDBK and HEK293A cells, respectively, and stored at −80 °C. All viruses and cells were obtained from the departmental collection. All biological assays, including virus cultivation and animal experiments with infectious virus particles were performed in the biosafety level 3 plus (BSL3+) and animal biosafety level 3 plus (ABSL3+) facilities of the Virology Department, Veterinary Faculty of Ankara University, Turkey.
2.3. Recombinant BoHV4-∆TK-CCHFV-N
CCHFV (Ank-2 strain) genomic RNA extraction was performed using the QIAamp Viral RNA Mini Kit (QIAGEN, Germantown, MD, USA) according to the manufacturer’s instructions. Superscript IV reverse transcriptase (Thermo Fisher Scientific) was employed for cDNA synthesis.
Table 1 shows the primer sets used to amplify the complete N coding gene of CCHFV based on the Turkey-Kelkit06 complete sequence (GenBank Accession Number: GQ337053), TK-CMV-N-TK homologous cassette to the thymidine kinase gene of the BoHV-4 Movar33/63 strain and the S gene PCR product containing the 50 bp homologous arms of the pCDNA3.1 myc/HisA vector. In the first step, the pCD-N1 construct was generated by the insertion of the gel-purified N PCR product between the EcoRI and XhoI sites of the pCDNA3.1 myc/His A plasmid (Invitrogen, Carlsbad, CA, USA). The final construct was verified by sequencing. In the next step, to propagate the TK-CMV-N-TK cassette from pCD-N1, primer sets (TM1 and TM2) containing the 50 bp homologous arms of the thymidine kinase gene (GenBank Accession Number: AF318573) of BoHV4-BAC (iBAC) were used. This cassette was used to perform recombineering in SW102 bacterial cells that previously contained BoHV4-loxp-BAC-CMV-EGFP-loxp (iBAC) following gel purification, according to the standard protocol (Available online:
https://redrecombineering.ncifcrf.gov/protocols/) [
21]. This bacterial strain is derived from DY380 (DH10B-derived strain) and, therefore, contains a defective λ prophage with the recombination proteins, exo, bet, and gam, under the control of the temperature-sensitive repressor,
cI857. To perform recombineering, the gel-purified TK-CMV-N-TK cassette (100 ng) was electroporated into heat-induced SW102 (42 °C for 15 min) containing iBAC in a 0.1-cm cuvette using a micropulser electroporator (BioRad, Hercules, CA, USA), and was subsequently plated on Luria–Bertani (LB) agar containing 12.5 µg/µL of chloramphenicol. Following the extraction of the verified BoHV4-BAC-∆TK-CCHFV-N DNA from SW102 by the alkaline lysis method, the MDBK cells were transfected using Lipofectamine 3000 reagent (Thermo Fisher Scientific) to generate infectious BoHV4-BAC-∆TK-CCHFV-N viruses. To eliminate loxp-BAC-EGFP-loxp from the final construct, recombinant BoHV4-BAC-∆TK-CCHFV-N was propagated in MDBK-cre cells to create BoHV4-∆TK-CCHFV-N viruses.
2.4. Recombinant Ad5-N
The AdMax™HI-IQ Kit J (MICROBIX Biosystems, Ontario, Canada) was used to generate Ad5-N. Basically, the S gene was cloned into the Ad5 shuttle vector (pDC516), which was flanked at 5′ by the Cytomegalovirus promoter (CMV) and at 3′ by an SV40 polyadenylation signal sequence, and contained E1 homologous arms to the pBHGfrt∆E1 plasmid. The seamless ligation cloning extract (SLiCE) method was used, which includes PCR amplification by primers targeting the S gene with the 50 bp homologous arms at the 5′ and 3′ sides of the EcoRI of the pDC516 vector, according to the standard protocol [
20].
Table 1 shows the employed primer set 1. The SLiCE extract was prepared from PPY bacteria (DH10B-derived
E. coli strain:F
− endA1
recA1
galE15
galK16
nupG
rpsLΔ
lacX74
Φ80lacZΔ M15
araD139Δ(
ara,leu)7697
mcrAΔ(
mrr-hsdRMS-
mcrBC)
cynX: [
araCpBAD
-redα EM7-
redβTn5-
gam]
λ−) as described elsewhere [
22]. The final construct (pDC516-N) was confirmed by colony PCR, restriction enzyme analysis and sequencing of the target gene. To rescue recombinant viruses, homologous recombination was achieved with the co-transfection of the pDC516-N (5 µg) and pBHGfrt∆E1 (3 µg) plasmids, using Lipofectamine 3000 reagent in a T25 cell culture flask containing HEK293 cells at a confluency of 90%. Twenty-four hours post-transfection, the cells were harvested by trypsin and transferred to a T75 culture vessel. The viruses were collected after 12–14 days. The cytopathic effects (CPEs), including rounded and detached cells, were then visualized in 90% of the monolayer. After 3 rounds of virus passage in HEK293 cells, a sufficient titer of the recombinant viruses was obtained. The recombinant viruses were titered in HEK293A cells by Adeno-X™ Rapid Titer Kit (Clontech, CA, USA). In addition, Ad5-wt virus was produced by the transfection of the pFG140 vector in HEK293 cells. Following titer determination, the virus stocks were stored at −80 °C.
2.5. In Vitro Detection of N Protein Expression
To confirm the in vitro expression of N proteins from the pCD-N1 and pDC516-N constructs, an indirect immunofluorescence assay (IIFA) was performed in BHK21-C13 cells, as previously described [
23]. Briefly, BHK21-C13 cells were cultivated in 24-well plates for 24 h before pCD-N1 transfection by Lipofectamine3000. The cells were fixed by 3.7% formaldehyde 48 h post-transfection, blocked with 5% skimmed milk (Cell Signaling, Leiden, The Netherlands) in 1x tris based buffer (TBS) buffer containing 0.2% Tween-20 (1xTBST), and incubated with a 1:250 dilution of primary human anti-CCHFV-N polyclonal antibodies in 1xTBST for 90 min at room temperature (RT). Then, Fluorescein isothiocyanate (FITC) -labeled anti-human IgG secondary antibody (Sigma) at a dilution of 1:750 was added and incubated for 1 h. The assay was evaluated by examining the cells with an Axio Vert A1 Microscope (Ziess, Oberkochen, Germany).
N protein expression in recombinant viruses was evaluated by Western blot assay. Infected MDBK (BoHV4-∆TK-CCHFV-S) and HEK293 (Ad5-N) cells (10 moi) were scraped 96 h post-inoculation and total proteins were extracted by the PRO-PREP™ protein extraction solution (iNtRON Biotechnology, Burlington, Massachusetts, USA), separated in Mini-Protean TGX Stain-Free precast gels (BioRad) in 1xTris/Glycine/SDS buffer and then transferred to a polyvinylidene difluoride (PVDF; BioRad) membrane using the Trans-Blot Turbo Transfer System (BioRad). The membrane was subsequently blocked with 5% skimmed milk in 1xTBST buffer for one hour followed by incubation with primary human polyclonal antibody at a dilution of 1:250 in 1xTBST for 2 hours at RT. Anti-human IgG-HRP secondary antibody (Sigma) at a dilution of 1:750 was added and incubated for an additional hour with gentle shaking. Finally, bands were visualized after the incubation of the membranes in a Clarity Western ECL substrate solution (BioRad) for 10 min in the dark and imaged using the ChemiDoc MP System (BioRad). Anti-Beta Actin antibody (St John’s Laboratory, London, UK) and CCHFV Ank-2-infected SW-13 cells (0.1 moi) at 3 days post-inoculation were used as controls.
2.6. Immunization Schedule of BALB/c Mice
A total of twenty-four female BALB/c (8–10 weeks old) mice were used in the study (
Table 2). The animals were randomly divided into six groups of four individuals, comprising BoHV4-∆TK-CCHFV-N, Ad5-N, Movar33/63, Ad5-wt, pCD-N1 and pCDNA3.1 myc/His A and a negative control (normal saline). At day 0, the study groups received 100TCID
50/0.3 mL doses of the relevant viruses through the intra-peritoneal route (i.p.), while the pCD-N1 and pCDNA3.1 myc/His A groups were injected with 50 µg of the constructs through the thigh muscle of the hind limb (i.m.). Booster injections were given after 2 weeks of the same regime. Serum samples were obtained from blood collected through the tail vein on days 0, 14 and 28, and stored at −80 °C until further analysis. In addition, splenocytes from individual mice were collected on day 28 for cytokine analysis.
2.7. Immunization and Challenge Experiment of IFNα/β/γR−/− Mice
The CCHFV Ank-2 strain, which was previously demonstrated to be lethal for IFNα/β/γR−/− mice, was used in the intra-peritoneal challenge experiment [
23]. The immunization schedule (vaccine constructs and their respective backbones) of IFNα/β/γR−/− mice (8–12-week-old female) was similar to BALB/c mice. All animals (4 animals/group) received 100TCID
50/300 µL of recombinant/wild type viruses and 50 µg/100 µL of DNA constructs (pCD-N1 and pCDNA3.1) on days 0 and 14. Serum samples were collected on days 0, 14, 28, prior to the challenge and stored at −80 °C to perform cytokine analysis and ELISA for the detection of N-specific IgG. To perform the challenge experiment, each group received 300 µL of a lethal dose of the virus (100LD
50 = 1000TCID
50; third passage in SW-13 cell) on day 28 and the assay continued for 13 days. Daily observations of clinical signs were recorded, including nasal or ocular discharge, appearance change, weight loss, depression and death. The euthanasia criteria for the surviving groups were based on complete recovery from clinical signs and stability of body weight for a minimum of 3 days.
2.8. Total Antibody Isotyping Assay from Immunized BALB/c Mice
A total antibody Isotyping ELISA kit (Thermo Fisher Scientific) was used to determine total immunoglobulin isotypes in immunized BALB/c mice serum samples collected on days 0 and 28, as previously described [
23].
2.9. ELISA from Serum Samples of Immunized IFNα/β/γR−/−
To detect the potential of our N-expressing constructs to induce specific antibody responses in IFNα/β/γR−/− mice (before the challenge experiment), we developed an in-house enzyme linked immunosorbent assay (ELISA). Briefly, infected SW-13 cells with 1 moi of the Ank-2 strain were collected 2 days post-inoculation (dpi) and cell lysates were used as ELISA antigens. A total of 45 µg of the cell lysate was added to each well of a Nunc MaxiSorp flat-bottom (Thermo Fisher Scientific) in a bicarbonate buffer (pH 8) and incubated overnight at 4 °C. The next day, 1/1000 diluted serum samples (on day 28) from immunized IFNα/β/γR−/− mice were added to each well and incubated for 2 h at room temperature (RT). Subsequently, anti-mouse IgG-HRP antibody (Abcam, Cambridge, MA, USA) at a dilution of 1/10,000 was added and further incubated at RT for 1 h, followed by adding 3, 3′, 5, 5′-Tetramethylbenzidine (TMB ELISA Peroxidase) substrate. Finally, the reaction was stopped by adding 2N H2SO4. The results of the unvaccinated mice serum samples were subtracted from the cell lysate as background.
2.10. Antibody Passive Transfer and T Cell Adoptive Transfer
Splenocytes and serum samples from immunized BALB/c mice were harvested on day 28 (two weeks after the booster dose). After staining with trypan blue (Thermo Fisher Scientific), 2 × 105 splenocytes combined with 100–300 µL of serum containing 500 µg of IgG antibody (measured by mouse anti-IgG kit, Wuhan Fine Biotech Co., Ltd. Wuhan, China) were injected through the intra-peritoneal route into IFNα/β/γR−/− mice (each group contained four 8- to 12-week-old female mice). Twenty-four hours later, the mice were challenged by a lethal dose (1000TCID50/300 µL) of the Ank-2 strain (i.p. route). The challenged animals were observed daily for onset of clinical signs and death. Survival rates and body weight percentages were recorded for a maximum of 15 days.
2.11. Virus Neutralization Assay (VNA)
A VNA was employed to elucidate the neutralizing ability of the actively immunized animals. Actively immunized BALB/c and IFNα/β/γR−/− mice serum samples, on days 14 and 28, were heat-inactivated at 56 °C for 30 min in a water bath and then serially diluted (1/2) in Dulbecco’s Modified Eagle’s medium (DMEM) and mixed with an equal volume of 100TCID50 of the virus in duplicate. After 1 h of incubation at 37 °C, the serum–virus mixture was inoculated in 1-day-old SW-13 cells at 90% confluency prepared in 24-well plates. The infected cells were further incubated under the same conditions for 4–5 days, with daily observation via an inverted microscope for virus-induced cytopathic effects.
2.12. Cytokine Assays
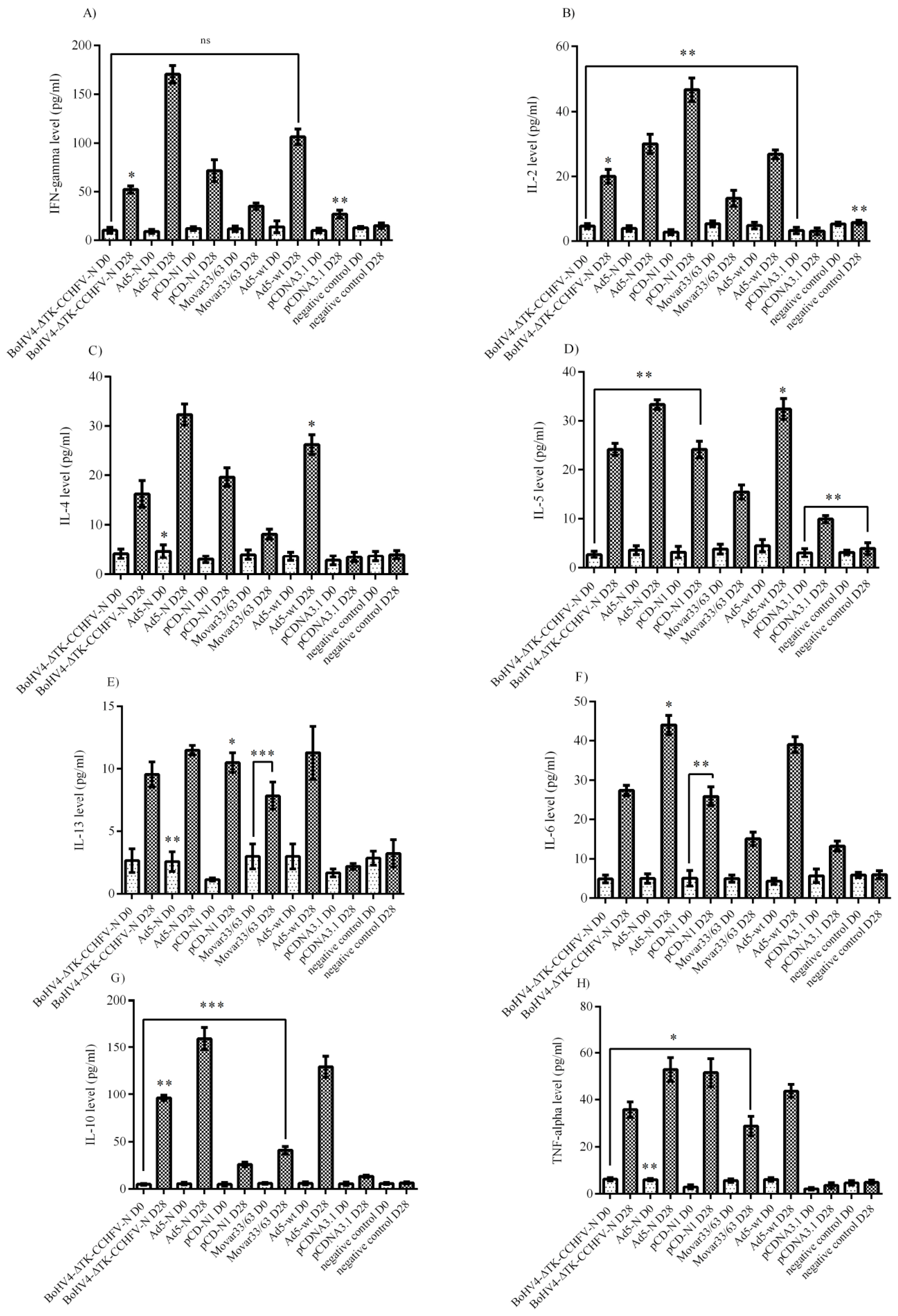
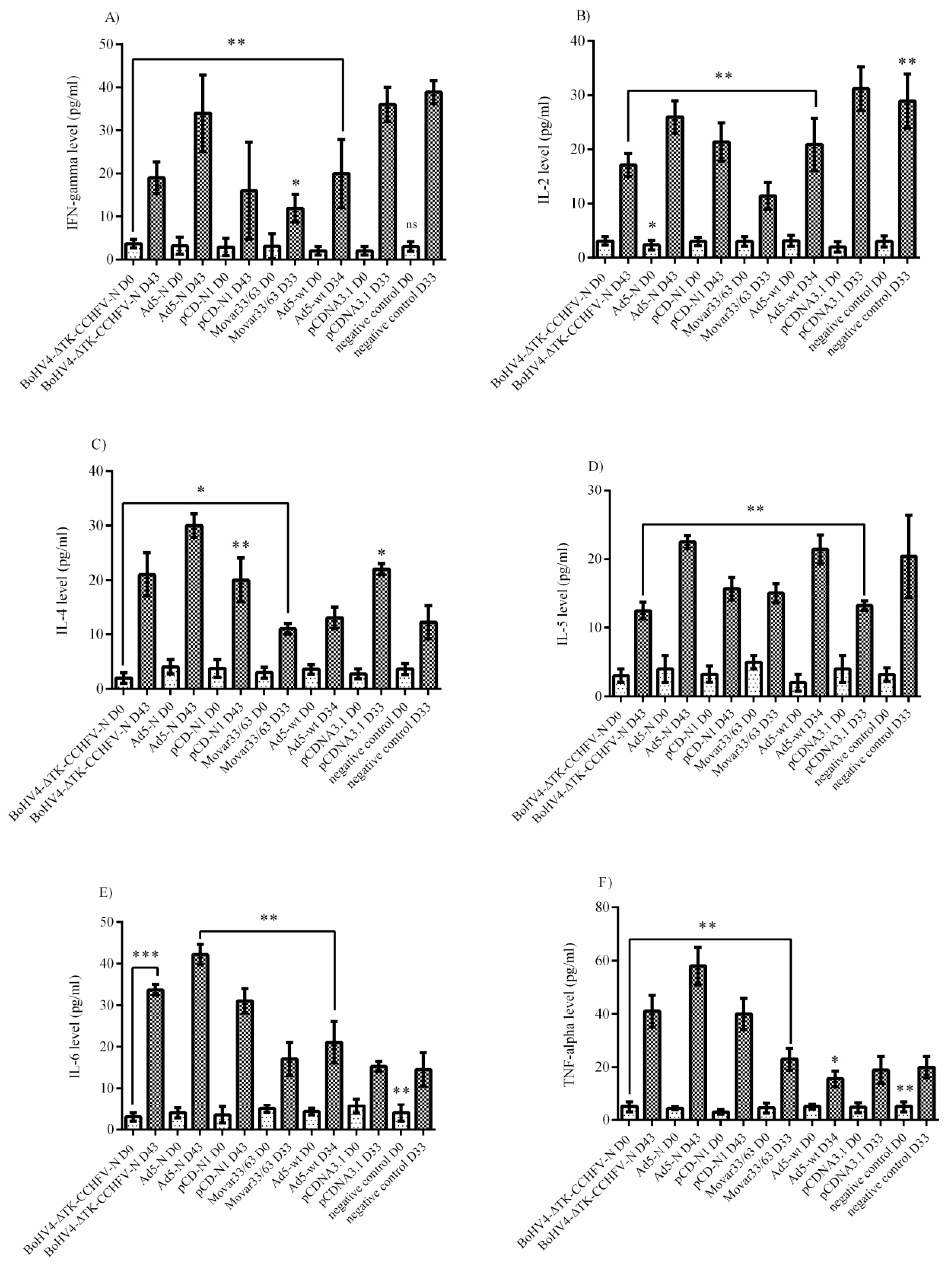
The supernatants collected from the virus-stimulated splenocytes of BALB/c and serum samples of BALB/c (on days 0 and 28) and IFNα/β/γR−/− (on days 0 and 33/34 for control groups/42 for survived groups from N-expressing groups) mice were subjected to cytokine measurements. To measure cytokine responses from virus-stimulated splenocyte supernatants, immunized BALB/c mice were euthanized on day 28 and their spleens were aseptically removed to prepare single cell splenocytes. After cell dissociation, red blood cells were lysed by red blood cell (RBC) lysis buffer (Biological Industries, Kibbutz Beit-Haemek, Israel) and the cells were resuspended in RPMI-1640 media (Sigma) then cultured in 24-well plates (250 × 103 per well). Immediately, 10 moi of Ank-2 strain (100 µL) was added to each well and the supernatants were collected after 48 and 72 h post-infection. The quantitation of cytokines from splenocyte supernatants (BALB/c) and serum samples (BALB/c and IFNα/β/γR−/−) was performed using the LEGENDplex™ Mouse Th1/Th2 Panel 8-plex kit (BioLegend, San Diego, CA, USA) by FacsCanto II Flow Cytometer (BD Bioscience, Franklin Lakes, New Jersey, USA). The results were analyzed by LEGENDplex™ data analysis software based on the manufacturer’s instructions.
5. Discussion
In parallel with the incidence of tick-borne pathogens, there is evident expansion of CCHFV activity, with the virus being reported from several previously unaffected regions [
24]. Due to the lack of a licensed vaccine or effective therapeutics, the development of new approaches to ensure protective immunity to CCHFV in susceptible individuals has become imperative. Previous efforts to develop immunity in indigenous populations of endemic regions, such as the Russian/Bulgarian vaccine construct, which contains chloroform-inactivated virus heated at 58 °C and absorbed on Al(OH)
3, failed due to various drawbacks [
25]. The main obstacle of the vaccine which was thoroughly evaluated for its efficacy in 2012 is the need for several booster doses to induce sufficient IFN-gamma response in volunteers [
26]. In addition to employing inactivated viruses, various strategies have been explored for an effective CCHFV vaccine development such as targeting viral glycoproteins and the nucleocapsid using several DNA or virus-like particle-based vaccines [
6,
27,
28].
In the present study, we set out to focus on the N protein for use in three different immunization platforms, namely, the adenoviral (Ad5-N) vector and a newly developed Bovine herpesvirus type 4 viral (BoHV4-∆TK-CCHFV-N) vector, alongside a DNA-based plasmid (pCD-N1). The CCHFV nucleocapsid protein (N), an essential component for intracellular virus replication, provides a prominent target for vaccine development as it is well conserved among global viral lineages and carries T cell epitopes. It was previously shown that the N-based vaccines of closely related viruses such as
Hantavirus and Rift Valley fever virus had the potential to elicit sufficient immune responses for protection in challenge assays [
29,
30,
31]. In addition, almost all vaccine development efforts based on the N protein of CCHFV have observed a protective rate of 100% in different expressing platforms and knock-out mice models [
7,
8,
23,
27].
The safety and flexibility of the viral vectors make them an interesting choice in both vaccination and oncolytic therapies. However, due to some obvious drawbacks of routinely used viral vectors, studies to develop new gene transfer systems are one of the most appealing research areas. For instance, a major obstacle of adenovirus-based vectors is the pre-existing immune response, which may cause inefficient expression following human vaccination. Employing rare virus serotypes, such as 2 and 5, as performed in this study, may overcome or alleviate this problem. In addition, adenovirus-based vectors are also highly immunogenic, so booster doses in individuals may induce serious side effects [
32].
One of the crucial requirements of newly developed vaccine vectors is their capability to efficiently express genes of interest to specifically stimulate immune responses in the target animal model. It has previously been demonstrated that the BoHV-4-based viral vector can stimulate immune responses when expressing immunogenic dominant antigens of diverse viral diseases such as Ebola and Bovine herpesvirus type 1 [
11]. For the first time, we have used this viral vector expressing an antigen from the
Bunyaviridae family to examine its immunological potential against the CCHFV N protein in BALB/c and IFNα/β/γR−/− mice models. In addition, we developed two other constructs expressing the same antigen (Ad5-N and pCD-N1) to evaluate the superiority of the BoHV-4-based vaccine over them. Following the in vitro generation of the vaccine constructs, we immunized BALB/c and IFNα/β/γR−/−mice and the immune responses were analyzed via cytokine assays in virus-stimulated splenocytes (BALB/c) and serum samples (BALB/c and IFNα/β/γR−/−) and total/specific immunoglobulin subtypes in serum samples (BALB/c and IFNα/β/γR−/−). The neutralization potential of the immunoglobulins produced was further assessed via virus neutralization assays in both animal models. We observed that N-expressing systems (BoHV4-∆TK-CCHFV-N, Ad5-N and pCD-N1) could induce prominent cytokine responses in both BALB/c and IFNα/β/γR−/− mice after a two-dose injection of the vaccine constructs. In the BALB/c mice model, Ad5-N dominated in all cytokine responses over the other expression constructs in the supernatant of the virus-stimulated splenocytes. In addition, the BoHV4-∆TK-CCHFV-N construct further resulted in elevated IL-4, IL-6 and IL-10 responses in the same assay. The only significantly induced cytokine via pCD-N1 was IFN-gamma. On the other hand, the pCD-N1 construct showed high potential for IL-2, IL-4, IL-5, IL-13, IL-6 and TNF-alpha stimulation in the BALB/c mice serum samples. For a deeper investigation of the protective mechanism involved in the lethal challenge, we analyzed cytokine responses in IFNα/β/γR−/− mice serum samples on days 0, 28 (2 weeks after booster and before challenge) and different days post-challenge (death days in control mice and day 43 for the survivor groups). We observed a negligible potential of BoHV4-∆TK-CCHFV-N, Ad5-N and pCD-N1 to stimulate IFN-gamma, IL-2 and IL-5. In contrast, all three constructs showed high levels of IL-6 and TNF-alpha, which can be considered as associated with survival in the challenged mice.
To highlight the strength of the antibody responses in the recovered human patients, it is worth mentioning that IgM and IgG are serological markers of virus exposure that become detectable during or after the first week of the infection in the survivors. In fatal cases, a lack of IgG and IgM responses has been documented, indicating the impact of antibodies (although non-neutralizing) in the immune control of the virus replication [
28,
33,
34]. In the serological assays of our study, the BoHV4-∆TK-CCHFV-N, Ad5-N and pCD-N1 constructs triggered the evaluated amount of total IgG immunoglobulin subtypes in BALB/c mice serum samples. We further evaluated the specific IgG antibody responses in IFNα/β/γR−/− mice (on day 28 and before the challenge experiment) by developing an in-house ELISA assay using CCHFV-infected cells and observed that pCD-N1 possessed more potential for humoral response stimulation. The other constructs also elicited significantly different IgG isotypes levels on day 28 (2 weeks after the second injection) that were analyzed by this assay. However, the total and specific antibody responses elicited by our N-expressing constructs failed to neutralize CCHFV in vitro, as assessed by the virus neutralization assay. The inability of the N protein to produce sufficient amounts of specific neutralizing antibodies was previously documented in knock-out animal models while non-neutralizing antibodies were also reported to protect mice from lethal CCHFV challenges [
23,
35,
36].
In addition, in this study, we showed the role of T cell and passive antibody transfer of immunized BALB/c mice in CCHFV lethal challenge protection in IFNα/β/γR−/− mice models. As previously shown, balanced Th1 and Th2 responses are necessary to elicit protection in challenge experiments, so we decided to try a combination of T cell and antibody in this assay [
8]. Antibody plus T cells of BoHV4-∆TK-CCHFV-N-immunized BALB/c mice elicited a 75% protection rate in IFNα/β/γR−/− mice during the challenge assay. This assay was previously conducted in the vaccinated mice by modified vaccinia Ankara expressing glycoprotein (MVA-GP), although no protection was documented [
37].
Our findings indicate that, despite the variation between cytokine and antibody responses in BALB/c mice, immunization with the CCHFV-N-expressing BoHV-4 viral vector has a significant protection potential against lethal challenge in both immunized and T cells/antibody passive transfer IFNα/β/γR−/− mice. However, due to the similar protection rates observed in BoHV4-∆TK-CCHFV-N and Ad5-N, a detailed comparison of these platforms could not be performed. Further experimental evaluation of different aspects of the immune response, such as CD4 and CD8 T cells, is therefore needed. Our clinical findings following challenge further suggest that Ad5-N induced more immunity, which caused milder symptoms, leading to faster recovery in the IFNα/β/γR−/− mice. Due to some advantages of the BoHV-4-based viral vector over the DNA and viral vectors, this viral vector can be considered predominant in circumstances such as those in this study, in which all the constructs had the potential to sufficiently stimulate an immune response to protect against the lethal challenge [
15]. One of the most important characteristics of BoHV-4 is its persistence in macrophages and monocyte cells. This may result in eliminating booster dose requirements due to frequent virus reactivation in persistence sites. In addition, macrophages and monocytes act as antigen-presenting cells, which can facilitate the presentation of antigens carried by the vector. However, the probable suppression of insert expression due to persistency resulting in insufficient immune stimulation must also be considered in the case of herpes viral vectors, including ours. The lack of a pre-existing immune response in humans and neutralizing antibodies in cattle, the main hosts of the virus, constitute additional advantages of BoHV-4-based delivery [
15]. Therefore, BoHV-4 should be explored in detail as a new potential viral vector for CCHFV vaccination.
In conclusion, our findings indicate that the CCHFV N protein can be considered as the main target in vaccine development regardless of the expressing platform. We also established a new viral vector expressing viral N protein (BoHV4-∆TK-CCHFV-N), which showed considerable potential as an antigen delivery system. The next step will be to focus on the immunological aspects of this newly developed platform in mice, or even in other animal models such as goat or sheep, which are considered to play important roles in virus epidemiology in nature.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}