1. Introduction
Lassa virus (LASV) has been listed by the World Health Organisation as one of the emerging pathogens likely to cause severe outbreaks in the near future and for which few or no medical countermeasures exist [
1]. In humans, it causes a viral haemorrhagic fever (VHF), Lassa fever (LF). LF is endemic in West Africa and causes tens of thousands of cases and several thousand deaths every year [
2]. There is currently no approved vaccine against LASV and only one partially efficient treatment, ribavirin [
3].
LASV is an old world arenavirus. This phylogenetic group includes highly pathogenic viruses, such as LASV or Lujo virus, and non-pathogenic viruses, such as Mopeia virus (MOPV) [
4]. MOPV is phylogenetically very close to LASV and has been isolated from
Mastomys natalensis, which is a LASV reservoir [
5]. However, MOPV is non-pathogenic in non-human primates (NHPs), and no human case of MOPV infection has ever been reported [
4]. Comparing MOPV and LASV, two very similar viruses with different pathogenic potential, may be a fruitful approach to identify immune and viral features involved in LASV pathogenesis. Fatal LASV infection in human is associated with immunosuppression, leading to uncontrolled viral replication and severe symptoms. In vivo studies have also shown that the survival of LASV-infected NHPs correlates with an early type I interferon (IFN-I) response and robust T-cell responses [
6].
Plasmacytoid dendritic cells (pDC) are highly potent IFN-I producers [
7]. They can detect viral infection through Toll-like receptors (TLR) 7 and 9, as well as RIG-I-like receptors (RLR). The constitutive expression of IRF7, which is usually an IFN-stimulated gene (ISG), partially explains the efficiency of IFN-I production by pDCs [
7]. During lymphocytic choriomeningitis virus (LCMV) infection, pDCs are the major source of IFN-I [
8]. In mice, pDCs were shown to be permissive to LCMV infection, and both infected and non-infected pDCs produced IFN-I [
9]. Considering these results and the major role of IFN-I in LF, we decided to study the response of pDCs to LASV. We compared LASV and MOPV to evaluate the importance of the pDC response in the high pathogenicity of LASV.
The results presented here were obtained using primary human pDCs purified from the blood of healthy donors. pDCs were not productively infected by either MOPV or LASV. On the contrary, coculture between pDCs and MOPV- or LASV-infected cells led to the detection of viral proteins in pDCs. We then analysed the IFN-I response of pDCs to MOPV and LASV. Both MOPV- and LASV-infected pDCs produced IFN-I. However, the response of pDCs to LASV was rapidly shut down. A larger scale approach, using transcriptomic and multiplex protein detection, showed stronger activation of MOPV-infected than LASV-infected pDCs. Overall, these results show that MOPV is a better stimulus for pDC activation than LASV. As pDCs are involved in the very early IFN-I response in vivo, their ability to detect LASV and initiate a response could be critical during LF.
2. Materials and Methods
2.1. Virus and Cells
VeroE6 cells were grown in DMEM with 0.5% penicillin-streptomycin (PS) and 5% foetal bovine serum (FBS, all from Invitrogen, Cergy-Pontoise, France). Mopeia (AN21366 strain [
4]) and Lassa (AV strain [
10]) viruses were grown in VeroE6 cells at 37 °C, with 5% CO
2. LASV and MOPV titres were determined by plaque immunoassays, as previously described [
11]. MOPV and LASV with a FLAG-tagged Z protein (MOPV-Zflag and LASV-Zflag) were obtained by reverse genetics, as previously described [
12]. Experiments with LASV were carried out in biosafety level 4 facilities (Laboratoire P4 Jean Merieux-Inserm, Lyon, France).
2.2. Cell Purification
Human peripheral blood was obtained from healthy donors with informed consent and was provided by the Etablissement Français du Sang (Lyon, France, agreement PLER/1-1820-05/05/14). Written informed consent was provided by all study participants. PBMCs were isolated by Ficoll (GE Healthcare, Velizy, France) centrifugation. pDCs were isolated using the Diamond Plasmacytoid Dendritic Cell Isolation kit II (Miltenyi Biotech, Paris, France). pDCs were cultured in RPMI 1640 Glutamax I, 0.5% PS, 10 mM HEPES, 1% nonessential amino acids, and 10% FBS (all from Invitrogen). R848 (Invitrogen) treatment at 1 µg/mL was used as a positive control of activation.
2.3. RT-qPCR
Cellular RNA was purified using the RNeasy kit (Qiagen, Courtaboeuf, France), followed by DNAse I (Qiagen) and Ambion DNAse (Thermo Fisher Scientific, Waltham, MA, USA) digestion. For viral RNA quantification, cells were treated prior to extraction with 0.05% trypsin (Invitrogen) to remove adsorbed virions. Viral RNA from the cell medium was purified using the QIAmp Viral RNA Mini Kit (Qiagen). IFN-I mRNA and viral nucleoprotein (NP) RNA was quantified by RT-qPCR, as previously described [
11]. Relative IFN-I mRNA levels were calculated as 2
−ΔCt, with Ct the cycle threshold and ΔCt = [gene Ct] − [GAPDH Ct]. NP RNA was quantified (by copy number) by comparing our samples with sequential dilutions of RNA standards. All runs were performed in duplicate using a LightCycler480 (Roche Diagnostics, Meylan, France).
2.4. Flow Cytometry
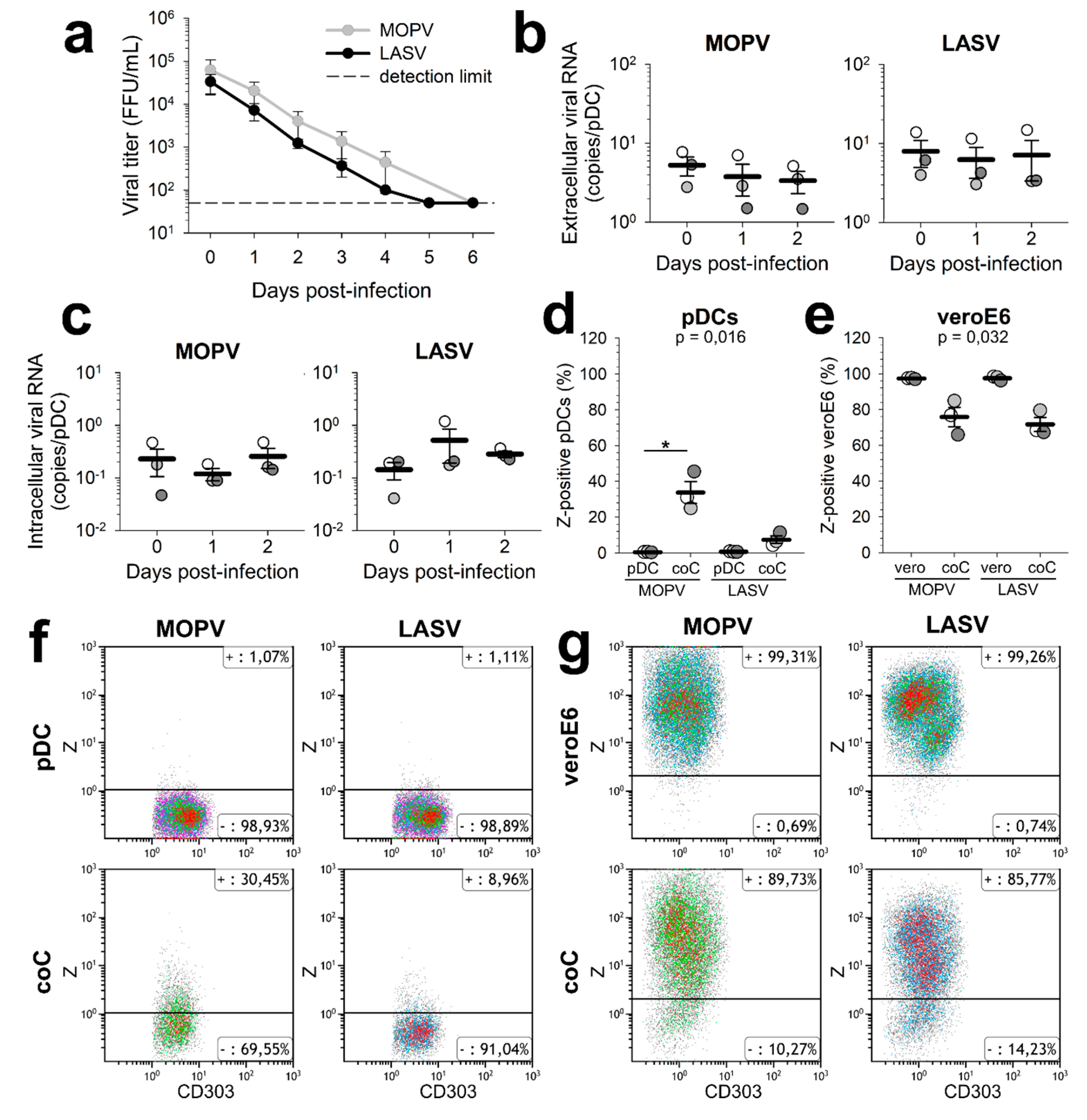
For the “vero” condition, VeroE6 cells were infected with MOPV-Zflag or LASV-Zflag (MOI = 0.3) and analysed 48 hours post infection (hpi) For the “pDC” condition, pDC were infected with MOPV-Zflag or LASV-Zflag (MOI = 0.1) and analysed 24 hpi. For the “coC” condition, VeroE6 cells were infected with MOPV-Zflag or LASV-Zflag (MOI = 0.3), pDCs were added 24 hpi and cells were analysed 48 hpi. For analysis, cells were stained with Lin1-FITC (BD Biosciences, Le-Pont-de-Claix, France) and CD303 (AC144)-PE-Vio770 (Miltenyi Biotech). Viral Z proteins were stained using the FoxP3 Staining Buffer Set, FcR Blocking Reagent, and human and anti-DYKDDDDK-APC (Miltenyi Biotech). Fluorescence was measured using a Gallios flow cytometer (Beckman Coulter, Brea, CA, USA) and analysed using Kaluza software version 1.2 (Beckman Coulter). pDCs were gated as Lin1-/CD303+ cells (
Figure S1).
2.5. Transcriptomic Analysis
pDCs were infected for 12 h at a MOI = 1 with LASV, MOPV, or remained uninfected. Cellular RNA was purified using the RNeasy kit (Qiagen), followed by DNAse I (Qiagen) and DNAse (Ambion) digestion. Sequencing were performed by ViroScan3D (Lyon, France). RNA quality was checked using QuantiFluor RNA System (Promega, Charbonnières-les-Bains, France) and RNA 6000 Pico Kit (Agilent, Santa Clara, CA, USA). cDNA were synthesized using random priming of poly-A RNAs (NEXTFLEX Rapid Directional RNA-Seq Library Prep Kit, PerkinElmer, Boston, MA, USA). Single-end, 75-bp read-length NextSeq 500 High throughput sequencing of the cDNA library was performed. After demultiplexing and trimming of the adaptors (with Bcl2fastq), 30 million reads per sample were obtained. Sequencing quality was assessed for each sample (before and after mapping) using FastQC. Reads were aligned on the human genome (Human GRCh38.p7, from ENSEMBL) using STAR and a maximum mismatch rate of 5%. Reads aligned on each gene were counted using the module feature count. Statistical analysis of the read counts (quality checks, normalisation using scaling factors, fold change and
p-values calculations) were performed using the R package SARTools (DESeq2) [
13]. Genes were differentially expressed for
p < 0.05. Heatmaps were generated with R (package heatmap2), using genes with differential expression for at least one pairwise comparison.
2.6. Luminex
pDCs were harvested 16 hpi and the culture medium collected. Fifty cytokines were quantified using the Milliplex map kit Human Cytokine/Chemokine Magnetic Bead Panel (PX38) and Human Cytokine/Chemokine Magnetic Bead Panel IV (Merck Millipore, Guyancourt, France). Runs were performed with a Magpix luminex (Merck Millipore).
2.7. Statistical Analysis
The mean and standard error of the mean (SEM) for each set of data were calculated using R. Graphs were generated and statistical analysis performed using SigmaPlot (SyStat Software Inc, San Jose, CA, USA). Differences were considered statistically significant at p < 0.05. Heatmaps were generated using R.
4. Discussion
We first showed that neither MOPV nor LASV infection of pDCs was productive. However, dendritic cells (DCs) express large amounts of α-dystroglycan, which is the MOPV and LASV receptor. DCs also express the alternative LASV receptors ALIX, TIM-1 and DC-SIGN. Finally, LCMV pseudo-particles with LASV glycoprotein have shown a great affinity for pDCs [
9]. Consequently, MOPV and LASV viral particles should be able to enter pDCs, although we do not know which receptors are expressed by human pDC. The viral cycle may have been initiated and aborted. We did not detect the production of MOPV and LASV RNAs in pDCs either. Therefore, if the viral cycle is initiated, it is aborted before the viral replication and transcription steps. As pDCs are highly efficient in pathogen detection, it is also possible that viral components were produced but quickly recycled, preventing their detection with our techniques. Studies on the infection of pDC-like cell lines by LCMV have shown no permissivity to viral particles. However, LCMV infection was possible through cellular contact with infected cells [
15]. With our model, we were able to detect MOPV and LASV Z proteins inside pDCs cultured with infected VeroE6 cells. These Z proteins could have three origins: neosynthesis, internalized virions or other type of viral components released by infected VeroE6 such as exosomes. Thus, coculture with infected cells either increased pDC permissivity to MOPV and LASV infection or increased the internalisation of viral component by pDCs. Further investigations using transwell experiments could allow to determine whether direct cell-cell contacts are needed for viral transmission to pDC. Depending on its origin, the Z protein could induce or elude different kinds of recognition pathways, which could affect pDC activation. We also showed that coculture with pDCs lowered the MOPV and LASV infection of VeroE6 cells. Production of IFN-I by pDCs could explain the control of VeroE6 infection, suggesting that pDCs respond to both MOPV- and LASV-infected VeroE6.
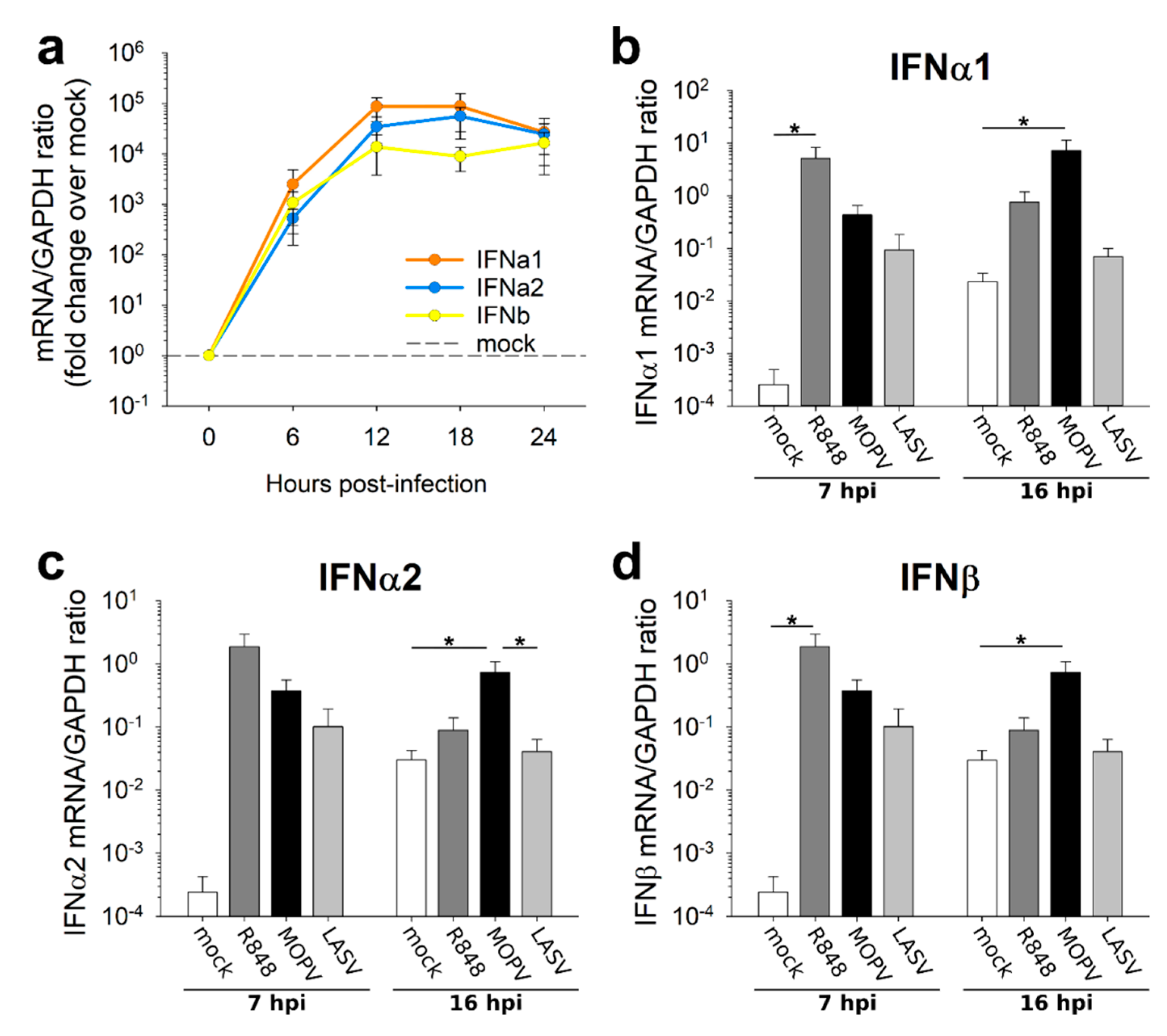
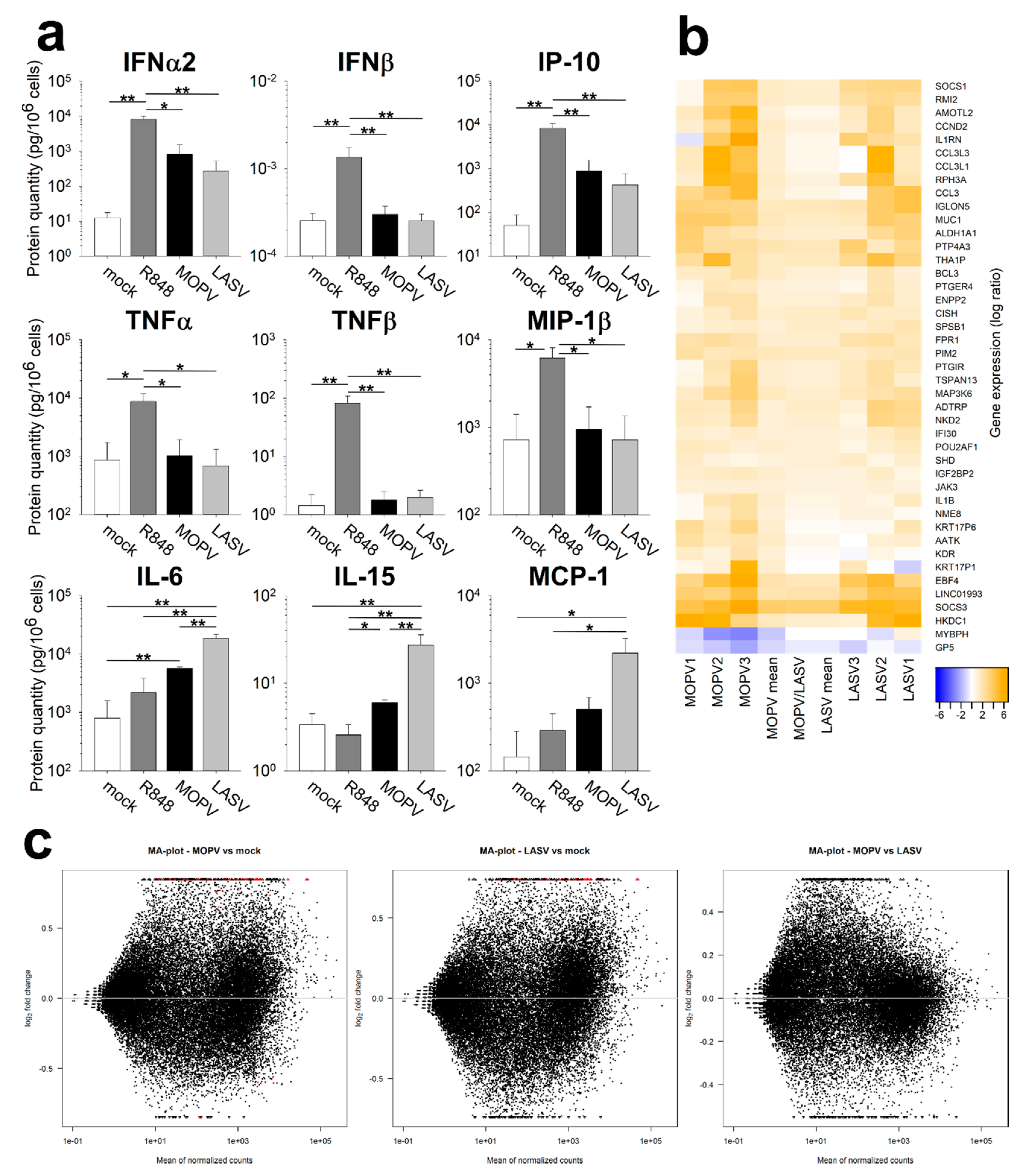
As pDCs are specialized in IFN-I production, we quantified their IFN-I response to MOPV and LASV. pDCs produced IFN-I in response to MOPV and LASV virions. However, pDC response to MOPV seemed to be more stable over time. This difference may rely on stronger activation by MOPV, allowing a longer response. It could also be explained by the expression of LASV proteins, which have immunosuppressive properties and may shut down the pDC response [
12]. Specifically, LASV NP has been shown to have major immunosuppressive properties in myeloid dendritic cells [
11]. However, we did not have the opportunity to evaluate the expression of NP in VeroE6-pDC cocultures. pDCs express a wide range of cellular sensors that can detect viral components. It may be informative to identify which pathways are responsible for MOPV and LASV sensing. Some LCMV strains, which are not detected by TLR7, do not induce IFN-I production by pDCs, suggesting that the pDC response to LCMV is TLR7 dependent [
7]. However, in vitro, RIG-I is also able to detect LASV and induces IFN-I production [
16]. MOPV and LASV may be detected by different sensors, influencing the pDC response.
To improve our understanding of pDC activation during MOPV and LASV infection, we quantified the cytokines produced by pDCs. IFN-I production by MOPV- and LASV-infected pDCs was confirmed. However, the absence of TNFα, TNFβ, and MIP-1β production suggests that MOPV- and LASV-infection did not lead to the complete activation of pDCs, in contrast to that induced by the TLR7 and TLR8 ligand R848. Unexpectedly, we showed that IL-6, IL-15, and MCP-1 were produced in higher amounts by LASV-infected than MOPV-infected pDCs. These three cytokines were previously identified in humans or NHPs with VHF. IL-6 was associated with severe cases of LASV and Ebola virus infection [
6,
17]. IL-15 and MCP-1 were also associated with a bad prognosis during the Crimean–Congo haemorrhagic fever [
18]. In pDCs, pro-inflammatory cytokines appear to be induced through IRF5 and NFκB activation [
19]. pDCs could be involved in the initiation of the pro-inflammatory context associated with severe LF, as the pDC response occurs very early. Transcriptomic analysis of MOPV- and LASV-infected pDCs suggested a comparable pattern of activation of pDCs between both viruses. However, MOPV infection had a greater impact on the pDC transcriptomic state compared to LASV infection. Therefore, MOPV seems to be a stronger stimulus than LASV for pDC activation.
Our results show that primary human pDCs are not productively infected by either MOPV or LASV. However, in the presence of infected cells, viral proteins could be detected in pDCs. MOPV-infected pDCs were globally activated, and rapidly and durably produced IFN-I. In contrast, LASV-infected pDCs were less activated, showed shorter IFN-I production, and produced pro-inflammatory cytokines. Further investigation will be necessary to identify the cell sensors involved in the response to LASV and MOPV and determine whether viral proteins are responsible for these different responses. The role of pDC in vivo could be critical, as IFN-I has been correlated with survival to LF. IFN-I produced by pDCs would have direct antiviral effects as well as induce better activation of other immune cells. Therefore, the strength of pDC activation during the very first steps of infection could modulate the global immune response to LASV.

 ,
, {kind=link}
{kind=link}
{kind=link}


