Development of a Versatile, Near Full Genome Amplification and Sequencing Approach for a Broad Variety of HIV-1 Group M Variants

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Clearance
2.2. Study Samples
2.3. RNA Extraction
2.4. cDNA Synthesis
2.5. Primer Design
2.6. PCR Amplification
2.7. Sanger Sequencing and Sequence Editing
2.8. Simplot and Recombinant Drawing Tool
2.9. Third-Generation Sequencing
2.10. Single-Gnome Amplification
2.11. Cloning
2.12. Data Storage and Documentation
3. Results
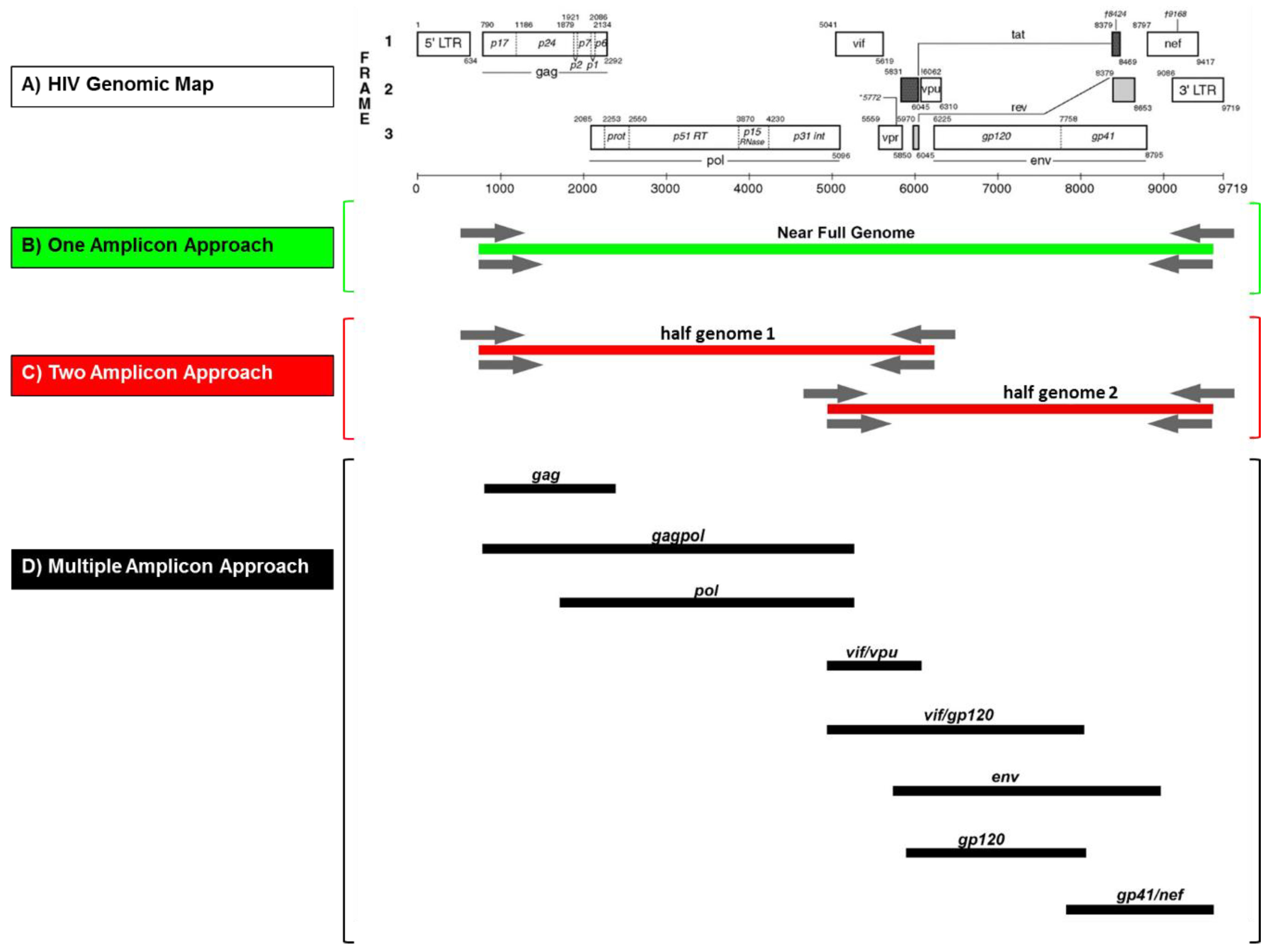
3.1. Design of an Adaptable NFG Amplification Strategy
3.2. Rational Primer Design for NFG Amplification and Sequencing of Diverse Group M Viruses
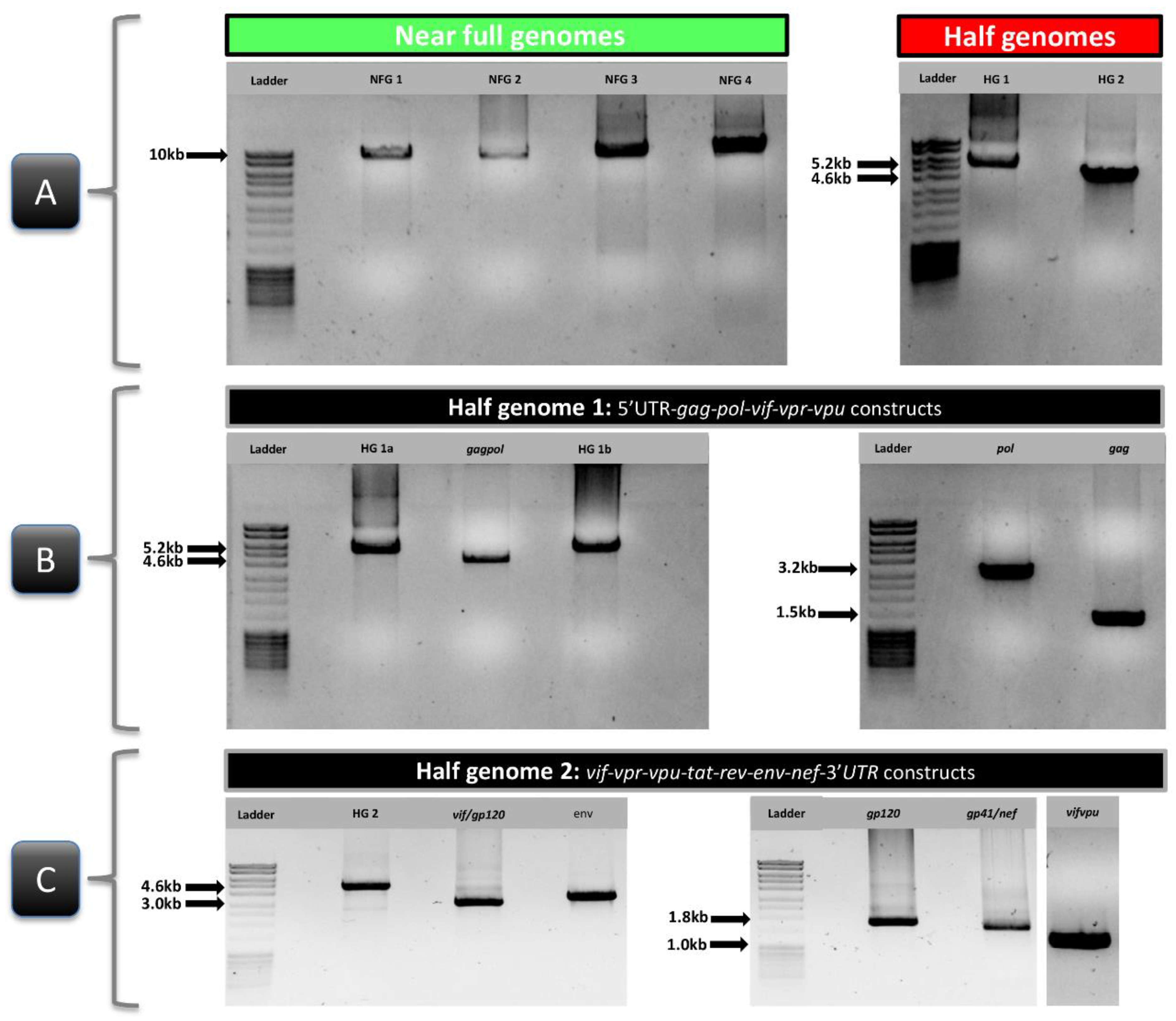
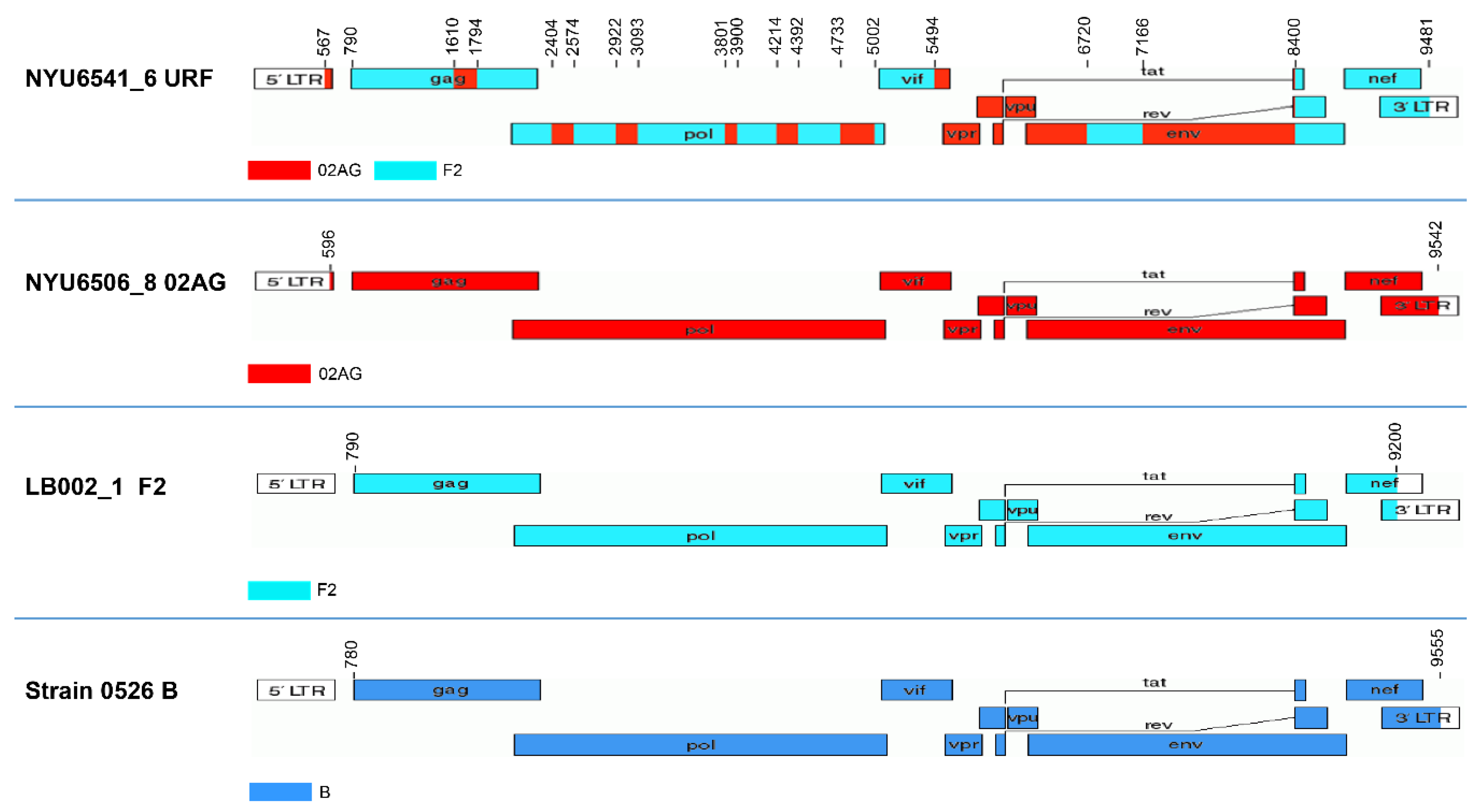
3.3. Subtype-Independent Amplification and Sequencing of HIV-1 Near Full Genomes
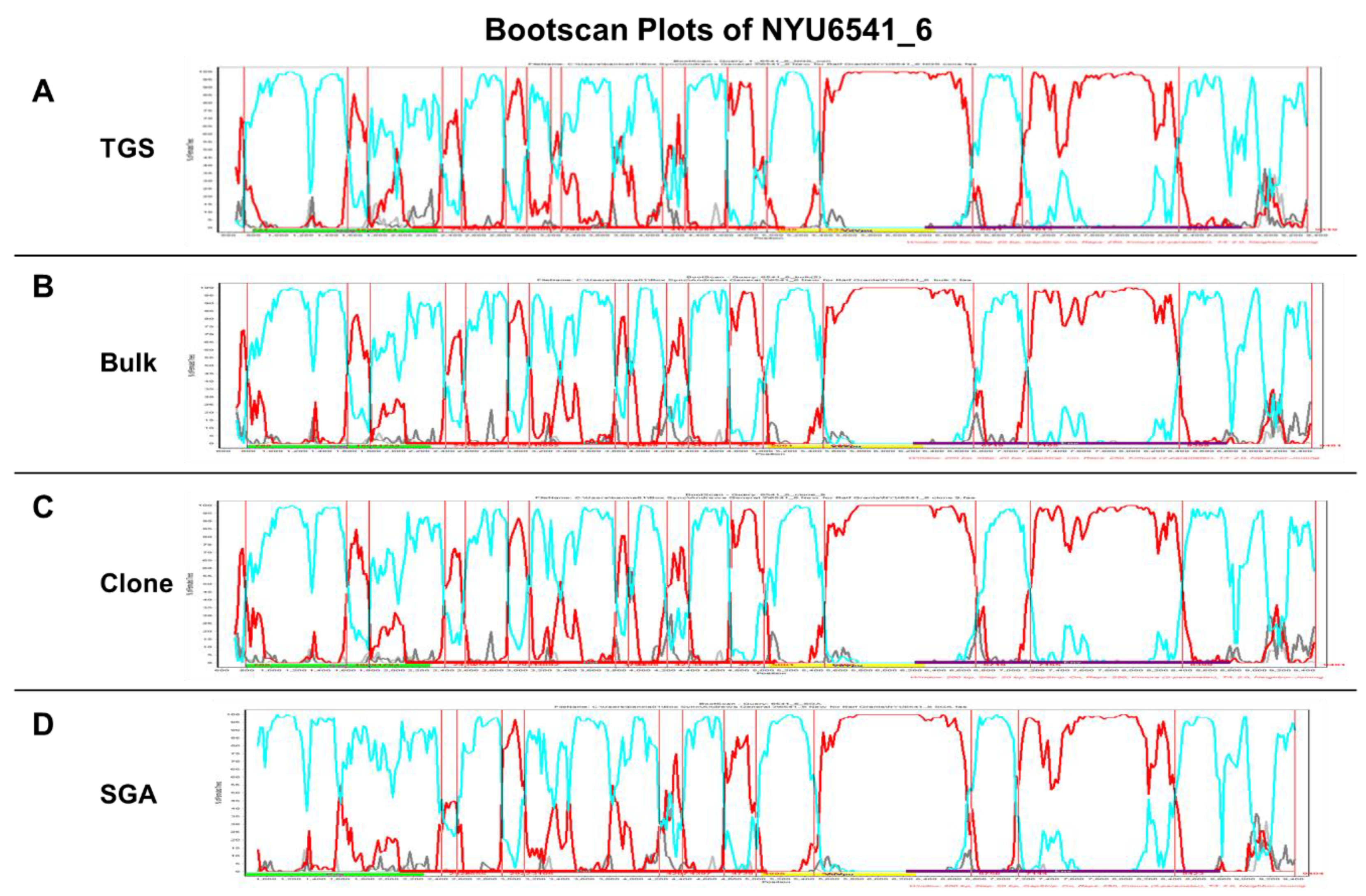
3.4. Comparative NFGS Analysis Using Different Amplification, Cloning, and Sequencing Strategies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gurtler, L.G.; Hauser, P.H.; Eberle, J.; von Brunn, A.; Knapp, S.; Zekeng, L.; Tsague, J.M.; Kaptue, L. A new subtype of human immunodeficiency virus type 1 (MVP-5180) from Cameroon. J. Virol. 1994, 68, 1581–1585. [Google Scholar]
- Plantier, J.C.; Leoz, M.; Dickerson, J.E.; de Oliveira, F.; Cordonnier, F.; Lemee, V.; Damond, F.; Robertson, D.L.; Simon, F. A new human immunodeficiency virus derived from gorillas. Nat. Med. 2009, 15, 871–872. [Google Scholar] [CrossRef]
- Vallari, A.; Holzmayer, V.; Harris, B.; Yamaguchi, J.; Ngansop, C.; Makamche, F.; Mbanya, D.; Kaptue, L.; Ndembi, N.; Gurtler, L.; et al. Confirmation of putative HIV-1 group P in Cameroon. J. Virol. 2011, 85, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Keele, B.F.; Van Heuverswyn, F.; Li, Y.; Bailes, E.; Takehisa, J.; Santiago, M.L.; Bibollet-Ruche, F.; Chen, Y.; Wain, L.V.; Liegeois, F.; et al. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science 2006, 313, 523–526. [Google Scholar] [CrossRef]
- HIV Circulating Recombinant Forms (CRFs). Available online: https://www.hiv.lanl.gov/content/sequence/HIV/CRFs/CRFs.html (accessed on 27 January 2019).
- Hemelaar, J.; Elangovan, R.; Yun, J.; Dickson-Tetteh, L.; Fleminger, I.; Kirtley, S.; Williams, B.; Gouws-Williams, E.; Ghys, P.D.; Characterisation, W.-U.N.f.H.I. Global and regional molecular epidemiology of HIV-1, 1990-2015: A systematic review, global survey, and trend analysis. Lancet Infect. Dis. 2018. [Google Scholar] [CrossRef]
- Reis, M.; Bello, G.; Guimaraes, M.L.; Stefani, M.M.A. Characterization of HIV-1 CRF90_BF1 and putative novel CRFs_BF1 in Central West, North and Northeast Brazilian regions. PLoS ONE 2017, 12, e0178578. [Google Scholar] [CrossRef]
- Villabona Arenas, C.J.; Vidal, N.; Ahuka Mundeke, S.; Muwonga, J.; Serrano, L.; Muyembe, J.J.; Boillot, F.; Delaporte, E.; Peeters, M. Divergent HIV-1 strains (CRF92_C2U and CRF93_cpx) co-circulating in the Democratic Republic of the Congo: Phylogenetic insights on the early evolutionary history of subtype C. Virus Evol. 2017, 3, vex032. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Ran, J.; Song, Y.; Liu, Y.; Gao, L.; Miao, Z.; Zhang, C.; Feng, Y.; Xia, X. Characterization of a Novel HIV-1 Circulating Recombinant Form, CRF01_AE/B’/C (CRF96_cpx), in Yunnan, China. AIDS Res. Hum. Retroviruses 2017. [Google Scholar] [CrossRef] [PubMed]
- Tongo, M.; Dorfman, J.R.; Martin, D.P. High Degree of HIV-1 Group M (HIV-1M) Genetic Diversity within Circulating Recombinant Forms: Insight into the Early Events of HIV-1M Evolution. J. Virol. 2015, 90, 2221–2229. [Google Scholar] [CrossRef]
- Hemelaar, J. Implications of HIV diversity for the HIV-1 pandemic. J. Infect. 2013, 66, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Foley, B.; Schultz, A.K.; Macke, J.P.; Bulla, I.; Stanke, M.; Morgenstern, B.; Korber, B.; Leitner, T. The role of recombination in the emergence of a complex and dynamic HIV epidemic. Retrovirology 2010, 7, 25. [Google Scholar] [CrossRef]
- HIV Sequence Database. Available online: https://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html (accessed on 27 January 2019).
- Smyth, R.P.; Schlub, T.E.; Grimm, A.J.; Waugh, C.; Ellenberg, P.; Chopra, A.; Mallal, S.; Cromer, D.; Mak, J.; Davenport, M.P. Identifying recombination hot spots in the HIV-1 genome. J. Virol 2014, 88, 2891–2902. [Google Scholar] [CrossRef]
- Fan, J.; Negroni, M.; Robertson, D.L. The distribution of HIV-1 recombination breakpoints. Infect. Genet. Evol. 2007, 7, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, C.M.; Birditt, B.A.; McKay, A.R.; Stoddard, J.N.; Lee, T.C.; McLaughlin, S.; Moore, S.W.; Shindo, N.; Learn, G.H.; Korber, B.T.; et al. Large-scale amplification, cloning and sequencing of near full-length HIV-1 subtype C genomes. J. Virol. Methods 2006, 136, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Nadai, Y.; Eyzaguirre, L.M.; Constantine, N.T.; Sill, A.M.; Cleghorn, F.; Blattner, W.A.; Carr, J.K. Protocol for nearly full-length sequencing of HIV-1 RNA from plasma. PLoS ONE 2008, 3, e1420. [Google Scholar] [CrossRef] [PubMed]
- Alampalli, S.V.; Thomson, M.M.; Sampathkumar, R.; Sivaraman, K.; U K J, A.J.; Dhar, C.; D Souza, G.; Berry, N.; Vyakarnam, A. Deep sequencing of near full-length HIV-1 genomes from plasma identifies circulating subtype C and infrequent occurrence of AC recombinant form in Southern India. PLoS ONE 2017, 12, e0188603. [Google Scholar] [CrossRef]
- Alves, B.M.; Siqueira, J.D.; Garrido, M.M.; Botelho, O.M.; Prellwitz, I.M.; Ribeiro, S.R.; Soares, E.A.; Soares, M.A. Characterization of HIV-1 Near Full-Length Proviral Genome Quasispecies from Patients with Undetectable Viral Load Undergoing First-Line HAART Therapy. Viruses 2017, 9, 392. [Google Scholar] [CrossRef] [PubMed]
- Henn, M.R.; Boutwell, C.L.; Charlebois, P.; Lennon, N.J.; Power, K.A.; Macalalad, A.R.; Berlin, A.M.; Malboeuf, C.M.; Ryan, E.M.; Gnerre, S.; et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 2012, 8, e1002529. [Google Scholar] [CrossRef] [PubMed]
- Gall, A.; Ferns, B.; Morris, C.; Watson, S.; Cotten, M.; Robinson, M.; Berry, N.; Pillay, D.; Kellam, P. Universal amplification, next-generation sequencing, and assembly of HIV-1 genomes. J. Clin. Microbiol. 2012, 50, 3838–3844. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.G.; Yamaguchi, J.; Alessandri-Gradt, E.; Tell, R.W.; Plantier, J.C.; Brennan, C.A. A Pan-HIV Strategy for Complete Genome Sequencing. J. Clin. Microbiol. 2016, 54, 868–882. [Google Scholar] [CrossRef]
- Heipertz, R.A., Jr.; Ayemoba, O.; Sanders-Buell, E.; Poltavee, K.; Pham, P.; Kijak, G.H.; Lei, E.; Bose, M.; Howell, S.; O’Sullivan, A.M.; et al. Significant contribution of subtype G to HIV-1 genetic complexity in Nigeria identified by a newly developed subtyping assay specific for subtype G and CRF02_AG. Medicine (Baltimore) 2016, 95, e4346. [Google Scholar] [CrossRef]
- Kijak, G.H.; Tovanabutra, S.; Sanders-Buell, E.; Watanaveeradej, V.; de Souza, M.S.; Nelson, K.E.; Ketsararat, V.; Gulgolgarn, V.; Wera-arpachai, M.; Sriplienchan, S.; et al. Distinguishing molecular forms of HIV-1 in Asia with a high-throughput, fluorescent genotyping assay, MHAbce v.2. Virology 2007, 358, 178–191. [Google Scholar] [CrossRef]
- Grossmann, S.; Nowak, P.; Neogi, U. Subtype-independent near full-length HIV-1 genome sequencing and assembly to be used in large molecular epidemiological studies and clinical management. J. Int. AIDS Soc. 2015, 18, 20035. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Olivo, A.; Laeyendecker, O.; Forberg, K.; Ndembi, N.; Mbanya, D.; Kaptue, L.; Quinn, T.C.; Cloherty, G.A.; Rodgers, M.A.; et al. Universal Target Capture of HIV Sequences from NGS Libraries. Front. Microbiol. 2018, 9, 2150. [Google Scholar] [CrossRef] [PubMed]
- Banin, A.N.; Tuen, M.; Tongo, M.; Bimela, J.S.; Nanfack, A.; Courtney, C.; Zappile, P.; Heguy, A.; Fokunang, C.; Mbanya, D.; Ngogang, J.; Nyambi, P.N.; Duerr, R. Near Full Genome Sequencing of Novel HIV-1 Unique Recombinant Forms Circulating in Cameroon. J. Int AIDS Soc. 2019. Submitted. [Google Scholar]
- Courtney, C.R.; Agyingi, L.; Fokou, A.; Christie, S.; Asaah, B.; Meli, J.; Ngai, J.; Hewlett, I.; Nyambi, P.N. Monitoring HIV-1 Group M Subtypes in Yaounde, Cameroon Reveals Broad Genetic Diversity and a Novel CRF02_AG/F2 Infection. AIDS Res. Hum. Retroviruses 2016, 32, 381–385. [Google Scholar] [CrossRef]
- Agyingi, L.; Mayr, L.M.; Kinge, T.; Orock, G.E.; Ngai, J.; Asaah, B.; Mpoame, M.; Hewlett, I.; Nyambi, P. The evolution of HIV-1 group M genetic variability in Southern Cameroon is characterized by several emerging recombinant forms of CRF02_AG and viruses with drug resistance mutations. J. Med. Virol. 2014, 86, 385–393. [Google Scholar] [CrossRef]
- Ragupathy, V.; Zhao, J.; Wood, O.; Tang, S.; Lee, S.; Nyambi, P.; Hewlett, I. Identification of new, emerging HIV-1 unique recombinant forms and drug resistant viruses circulating in Cameroon. Virol. J. 2011, 8, 185. [Google Scholar] [CrossRef]
- Salazar-Gonzalez, J.F.; Bailes, E.; Pham, K.T.; Salazar, M.G.; Guffey, M.B.; Keele, B.F.; Derdeyn, C.A.; Farmer, P.; Hunter, E.; Allen, S.; et al. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J. Virol 2008, 82, 3952–3970. [Google Scholar] [CrossRef] [PubMed]
- Tongo, M.; Dorfman, J.R.; Abrahams, M.R.; Mpoudi-Ngole, E.; Burgers, W.A.; Martin, D.P. Near full-length HIV type 1M genomic sequences from Cameroon: Evidence of early diverging under-sampled lineages in the country. Evol. Med. Public Health 2015, 2015, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef] [PubMed]
- Courtney, C.R.; Mayr, L.; Nanfack, A.J.; Banin, A.N.; Tuen, M.; Pan, R.; Jiang, X.; Kong, X.P.; Kirkpatrick, A.R.; Bruno, D.; et al. Contrasting antibody responses to intrasubtype superinfection with CRF02_AG. PLoS ONE 2017, 12, e0173705. [Google Scholar] [CrossRef]
- Mayr, L.M.; Powell, R.L.; Ngai, J.N.; Takang, W.A.; Nadas, A.; Nyambi, P.N. Superinfection by discordant subtypes of HIV-1 does not enhance the neutralizing antibody response against autologous virus. PLoS ONE 2012, 7, e38989. [Google Scholar] [CrossRef]
- Powell, R.L.; Urbanski, M.M.; Burda, S.; Nanfack, A.; Kinge, T.; Nyambi, P.N. Utility of the heteroduplex assay (HDA) as a simple and cost-effective tool for the identification of HIV type 1 dual infections in resource-limited settings. AIDS Res. Hum. Retroviruses 2008, 24, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Nanfack, A.J.; Redd, A.D.; Bimela, J.S.; Ncham, G.; Achem, E.; Banin, A.N.; Kirkpatrick, A.R.; Porcella, S.F.; Agyingi, L.A.; Meli, J.; et al. Multimethod Longitudinal HIV Drug Resistance Analysis in Antiretroviral-Therapy-Naive Patients. J. Clin. Microbiol 2017, 55, 2785–2800. [Google Scholar] [CrossRef] [PubMed]
- Alidjinou, E.K.; Deldalle, J.; Hallaert, C.; Robineau, O.; Ajana, F.; Choisy, P.; Hober, D.; Bocket, L. RNA and DNA Sanger sequencing versus next-generation sequencing for HIV-1 drug resistance testing in treatment-naive patients. J. Antimicrob. Chemother. 2017, 72, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef] [PubMed]
- Laver, T.; Harrison, J.; O’Neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 2015, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tyler, A.D.; Mataseje, L.; Urfano, C.J.; Schmidt, L.; Antonation, K.S.; Mulvey, M.R.; Corbett, C.R. Evaluation of Oxford Nanopore’s MinION Sequencing Device for Microbial Whole Genome Sequencing Applications. Sci. Rep. 2018, 8, 10931. [Google Scholar] [CrossRef] [PubMed]
- Aralaguppe, S.G.; Siddik, A.B.; Manickam, A.; Ambikan, A.T.; Kumar, M.M.; Fernandes, S.J.; Amogne, W.; Bangaruswamy, D.K.; Hanna, L.E.; Sonnerborg, A.; et al. Multiplexed next-generation sequencing and de novo assembly to obtain near full-length HIV-1 genome from plasma virus. J. Virol. Methods 2016, 236, 98–104. [Google Scholar] [CrossRef]
- Jordan, M.R.; Kearney, M.; Palmer, S.; Shao, W.; Maldarelli, F.; Coakley, E.P.; Chappey, C.; Wanke, C.; Coffin, J.M. Comparison of standard PCR/cloning to single genome sequencing for analysis of HIV-1 populations. J. Virol. Methods 2010, 168, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Sista, P.; Giguel, F.; Greenberg, M.; Kuritzkes, D.R. Relative replicative fitness of human immunodeficiency virus type 1 mutants resistant to enfuvirtide (T-20). J. Virol. 2004, 78, 4628–4637. [Google Scholar] [CrossRef] [PubMed]
- Parrish, N.F.; Wilen, C.B.; Banks, L.B.; Iyer, S.S.; Pfaff, J.M.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Decker, J.M.; Parrish, E.H.; Berg, A.; et al. Transmitted/founder and chronic subtype C HIV-1 use CD4 and CCR5 receptors with equal efficiency and are not inhibited by blocking the integrin alpha4beta7. PLoS Pathog. 2012, 8, e1002686. [Google Scholar] [CrossRef]
- Weber, J.; Vazquez, A.C.; Winner, D.; Rose, J.D.; Wylie, D.; Rhea, A.M.; Henry, K.; Pappas, J.; Wright, A.; Mohamed, N.; et al. Novel method for simultaneous quantification of phenotypic resistance to maturation, protease, reverse transcriptase, and integrase HIV inhibitors based on 3’Gag(p2/p7/p1/p6)/PR/RT/INT-recombinant viruses: A useful tool in the multitarget era of antiretroviral therapy. Antimicrob. Agents Chemother. 2011, 55, 3729–3742. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Portion Analyzed | Nested PCR Round | Primer ID | Sequence | Position in HxB2 | Temp. | Reference |
|---|---|---|---|---|---|---|
| Near full genome 1 (NFG1) | 1st Round | 5’ UTR_For1 | TGCCTTGAGTGCTTCAAGTAGTGTG | 536–560 | 58.4 °C | This study |
| 3’ UTR_Rev1 | TATTGAGGCTTAAGCAGTGGGTTC | 9615–9592 | 57 °C | This study | ||
| 2nd Round | 5’ UTR_For2 | TCTGTTGTGTGACTCTGGTAACTAGAGATC | 567–596 | 58.7 °C | This study | |
| 3’ UTR_Rev4 | GCTCAAATCTGGTCTAGCAAGAGAGA | 9567–9542 | 58 °C | This study | ||
| Near full genome 2 (NFG2) | 1st Round | HIV_682_For | TCTCTCGACGCAGGACTCGGCTTGCTG | 682–708 | 67 °C | [25] |
| HIV_9555_Rev | TCTACCTAGAGAGACCCAGTACA | 9555–9533 | 55.5 °C | [25] | ||
| 2nd Round | HIV_776_For | CTAGAAGGAGAGAGAGATGGGTGCGAG | 776–800 | 61 °C | [25] | |
| HIV_9555_Rev | TCTACCTAGAGAGACCCAGTACA | 9555–9533 | 55.5 °C | [25] | ||
| Near full genome 3 (NFG3) | 1st Round | HIV_682_For | TCTCTCGACGCAGGACTCGGCTTGCTG | 682–708 | 67 °C | [25] |
| 3’ UTR_Rev3 | AGAGCTCCCAGGCTCAAATCTGGTCTA | 9578–9552 | 62.5 °C | This study | ||
| 2nd Round | HIV_776_For | CTAGAAGGAGAGAGAGATGGGTGCGAG | 776–800 | 61 °C | [25] | |
| 3’ UTR_Rev4 | GCTCAAATCTGGTCTAGCAAGAGAGA | 9567–9542 | 58 °C | This study | ||
| Near full genome 4 (NFG4) | 1st Round | HIV_682_For | TCTCTCGACGCAGGACTCGGCTTGCTG | 682–708 | 67 °C | [25] |
| 3’ UTR_Rev5 | CAGTACAGGCGAGAAGCAGCTGCT | 9539–9516 | 62 °C | This Study | ||
| 2nd Round | HIV_776_For | CTAGAAGGAGAGAGAGATGGGTGCGAG | 776–800 | 61 °C | [25] | |
| 3’ UTR_Rev6 | AGCAGCTGCTTATATGCAGCATCTGAG | 9525–9499 | 60.7 °C | This Study | ||
| Half genome 1a (HG1a) | 1st Round | HIV_682_For | TCTCTCGACGCAGGACTCGGCTTGCTG | 682–708 | 67 °C | [25] |
| Vpu2_Rev | CCGCTTCTTCCTGCCATAGGA | 5985–5966 | 59.1 °C | This study | ||
| 2nd Round | HIV_776_For | CTAGAAGGAGAGAGAGATGGGTGCGAG | 776–800 | 61 °C | [25] | |
| Vpu3_Rev | TCCTGCCATAGGAGATGCCTAAG | 5978–5956 | 58.1 °C | This study | ||
| Half genome 1b (HG1b) | 1st Round | Gag3_For | GAGAGATGGGTGCGAGAGC | 785–803 | 57 °C | This study |
| Vpu2_Rev | CCGCTTCTTCCTGCCATAGGA | 5985–5966 | 59.1 °C | This study | ||
| 2nd Round | Gag4_For | TAGTATGGGCAAGCAGGGA | 890–908 | 56 °C | This study | |
| Vpu3_Rev | TCCTGCCATAGGAGATGCCTAAG | 5978–5956 | 58.1 °C | This study | ||
| Half genome 2 (HG2) | 1st Round | Vif2_For | TGGAAAGGTGAAGGGGCAGTA | 4956–4976 | 58.4 °C | This study |
| OFM19 | GCACTCAAGGCAAGCTTTATTGAGGCTTA | 9632–9604 | 60.9 °C | [31] | ||
| 2nd Round | Vif3_For | GATTATGGAAAACAGATGGCAGGT | 5037–5060 | 55.2 °C | This study | |
| HIV_9555_Rev | TCTACCTAGAGAGACCCAGTACA | 9555–9533 | 55.5 °C | [25] | ||
| gagpol | 1st Round | Gag2_For | GACTAGCGGAGGCTAGAAG | 764–782 | 54 °C | This study |
| Pol2_Rev | CCATGTTCTAATCCTCATCCTGTC | 5103–5080 | 55 °C | This study | ||
| 2nd Round | Gag3_For | GAGAGATGGGTGCGAGAGC | 785–803 | 57 °C | This study | |
| Pol3_Rev | CTGTCTACCTGCCACACA | 5084–5067 | 54 °C | This study | ||
| gag | 1st Round | Gag2_For | GACTAGCGGAGGCTAGAAG | 764–782 | 54 °C | This study |
| Gag1_Rev | CCAATTCCCCCTATCAT | 2404–2388 | 48 °C | This study | ||
| 2nd Round | Gag4_For | TAGTATGGGCAAGCAGGGA | 890–908 | 56 °C | This study | |
| Gag3_Rev | GGTCGTTGCCAAAGAGTGA | 2278–2260 | 55 °C | This study | ||
| pol | 1st Round | Pol1_For | GAAGAAATGATGACAGC | 1819–1835 | 45 °C | This Study |
| Pol1_Rev | TGCCAGTCTCTTTCTCCTG | 5279–5161 | 54 °C | This study | ||
| 2nd Round | Pol2_For | AAGTGTTTCAACTGTGG | 1960–1976 | 47 °C | This study | |
| Pol2_Rev | CCATGTTCTAATCCTCATCCTGTC | 5103–5080 | 55 °C | This study | ||
| vifvpu | 1st Round | Vif 1_For | GGGTTTATTACAGGGACAGCAGAG | 4900–4923 | 57.3 °C | [31] |
| Vpu1_Rev | TTGCCACTYTCTTCTGCTCTTTC | 6225–6203 | 56.4 °C | This study | ||
| 2nd Round | Vif 2_For | TGGAAAGGTGAAGGGGCAGTA | 4956–4976 | 58.4 °C | This study | |
| Vpu2_Rev | CCGCTTCTTCCTGCCATAGGA | 5985–5966 | 59.1 °C | This study | ||
| vif/gp120 | 1st Round | Vif1_For | GGGTTTATTACAGGGACAGCAGAG | 4900–4923 | 57.2 °C | This study |
| Gp120out | GCARCCCCAAAKYCCTAGG | 8018–8000 | 57.2 °C | This study | ||
| 2nd Round | Vif2_For | TGGAAAGGTGAAGGGGCAGTA | 4956–4976 | 58.4 °C | This study | |
| Gp120in | CGTCAGCGTYATTGACGCYGC | 7838–7818 | 61.4 °C | This study | ||
| env | 1st Round | Vif1_For | GGGTTTATTACAGGGACAGCAGAG | 4900–4923 | 57.2 °C | [31] |
| OFM19 | GCACTCAAGGCAAGCTTTATTGAGGCTTA | 9632–9604 | 60.9 °C | [31] | ||
| 2nd Round | Env A | (CACC)GGCTTAGGCATCTCCTATGGCAGGAAGAA | 5954–5982 | 63 °C | [31] | |
| 02AG-Env N | GTTCTGCCAATCTGGGAAGAATCCTTGTGTG | 9174–9144 | 62.3 °C | This study | ||
| gp120 | 1st Round | Env A | GGCTTAGGCATCTCCTATGGCAGGAAGAA | 5954–5982 | 62.8 °C | [31] |
| Gp120out | GCARCCCCAAAKYCCTAGG | 8018–8000 | 57.2 °C | [39] | ||
| 2nd Round | Env B | AGAAAGAGCAGAAGACAGTGGCA | 6202–6224 | 58.2 °C | [31] | |
| Gp120in | CGTCAGCGTYATTGACGCYGC | 7838–7818 | 61.4 °C | [39] | ||
| gp41/nef | 1st Round | Gp120in_For1 | CAGCAGGAAGCACTATGGGCG | 7798–7818 | 60.7 °C | This study |
| 3’UTR_Rev1 | TATTGAGGCTTAAGCAGTGGGTTC | 9615–9592 | 57 °C | This study | ||
| 2nd Round | Gp120in_For2 | GCRGCGTCAATRACGCTGACG | 7818–7838 | 61.4 °C | This study | |
| 3’UTR_Rev4 | GCTCAAATCTGGTCTAGCAAGAGAGA | 9567–9542 | 58 °C | This study |
| Primer ID | Sequence | Position in HxB2 | Temp. | Reference |
|---|---|---|---|---|
| HIV_580_For | TCTGGTAACTAGAGATCC | 580–597 | 46.6 °C | This study |
| HIV_625_For | ATCTCTAGCAGTGGCGCCCGA | 625–641 | 63 °C | This study |
| HIV_788_For | AGATGGGTGCGAGAGCGT | 788–809 | 59.2 °C | This study |
| HIV_1250_For | CATGGGTAAAGGTAATAGAAG | 1250–1270 | 47.9 °C | This study |
| HIV_1250b_For | CATGGGTAAAAGTAATAGAA | 1250–1269 | 44.5 °C | This study |
| HIV_1400_For | CCATCAATGAGGAAGCTGCA | 1400–1419 | 55.4 °C | This study |
| HIV_1830_For | TGATGACAGCATGCCAGG | 1830–1847 | 55.3 °C | This study |
| HIV_1970_For | TTCAACTGTGGCAAAGAAGG | 1970–1989 | 53.4 °C | This study |
| HIV_2075_For | GACAGGCTAATTTTTTAGGGA | 2075–2095 | 49.9 °C | This study |
| HIV_2590_For | CAGGAATGGATGGCCCAA | 2590–2607 | 55.2 °C | This study |
| HIV_2700_For | GGGCCTGAAAATCCATACAATACT | 2700–2723 | 55.1 °C | This study |
| HIV_2756_For | GTACTAAATGGAGAAAATTAG | 2756–2776 | 43.8 °C | This study |
| HIV_3300_For | AGCTGGACTGTCAATGA | 3300–3316 | 50.4 °C | This study |
| HIV_3350_For | GGGCAAGTCAAATTTATCCAG | 3350–3370 | 51.8 °C | This study |
| HIV_3355_For | AGCCAGATTTATCCAGG | 3355–3371 | 48.2 °C | This study |
| HIV_4000_For | TAGCCTTGCAGGATTCAGGAT | 4000–4020 | 56.0 °C | This study |
| HIV_4175_For | TGGAGGAAATGAACAAGTAGA | 4175–4195 | 51 °C | This study |
| HIV_4542_For | GCAGGAAGATGGCCAGT | 4542–4558 | 55.0 °C | This study |
| HIV_4646_For | TGGAATTCCCTACAATCC | 4646–4663 | 48.6 °C | This study |
| HIV_4747_For | AGACAGCAGTACAGATGGCAG | 4747–4767 | 56.7 °C | This study |
| HIV_4900_For | GGGTTTATTACAGGGACAGCA | 4900–4920 | 54.6 °C | This study |
| HIV_5388_For | TTTCAGAATCTGCCATAAG | 5388–5406 | 47.1 °C | This study |
| HIV_5769_For | CATTTCAGAATYGGGTG | 5769–5786 | 46.5 °C | This study |
| HIV_5840_For | GTAGATCCTARCCTAGA | 5840–5856 | 44.1 °C | This study |
| HIV_5970_For | ATGGCAGGAAGAAGCGGAGAC | 5970–5990 | 59.5 °C | This study |
| HIV_6100_For | AGTAGCATTCATAGCAGCCAT | 6100–6120 | 54.0 °C | This study |
| HIV_6125_For | GTGTGGACTATAGTATATATAG | 6125–6146 | 44.1 °C | This study |
| HIV_6380_For | ATTTTGTGCATCAGATGC | 6380–6397 | 44.7 °C | This study |
| HIV_6543_For | GATATAATTAGTCTATGGG | 6543–6561 | 40.8 °C | This study |
| HIV_6745_For | CACTTTTTTATAGACTTGAT | 6745–6764 | 42.7 °C | This study |
| HIV_6826_For | TTAMACAGGCTTGTCC | 6826–6841 | 47.2 °C | This study |
| HIV_6840_For | CCAAAGGTATCCTTTGAGCCA | 6840–6860 | 54.9 °C | This study |
| HIV_6855_For | GAGCCAATTCCCATACAT | 6855–6872 | 49.3 °C | This study |
| HIV_7500_For | ATGTGGCAGAAAGTAGGACAAGC | 7500–7522 | 57.1 °C | This study |
| HIV_7520_For | AGCAATGTATGCCCCTC | 7520–7536 | 52 °C | This study |
| HIV_7633_For | CTGGAGGAGGAGATATGAG | 7633–7651 | 50.7 °C | This study |
| HIV_7660_For | GGAGAAGTGAATTATATAA | 7660–7678 | 40.6 °C | This study |
| HIV_7803_For | GGAAGCACTATGGGCGC | 7803–7819 | 56.7 °C | This study |
| HIV_8800_For | GGTGGCAAGTGGTCAAA | 8800–8816 | 53.0 °C | This study |
| HIV_8180_For | GCAGGAAAAGAATGAACAAG | 8180–8199 | 49.7 °C | This study |
| HIV_8970_For | GGTTAGAAGCACAAGAG | 8970–8986 | 47.0 °C | This study |
| HIV_9015_For | GGTACCTTTAAGACCAATGA | 9015–9034 | 49.3 °C | This study |
| HIV_9030_For | GACCAATGACTTATAAGG | 9030–9047 | 44.1 °C | This study |
| Primer ID | Sequence | Position in HxB2 | Temp. | Reference |
|---|---|---|---|---|
| HIV_809_Rev | ACGCTCTCGCACCCATCT | 809–792 | 58.9 °C | This Study |
| HIV_912_Rev | TCCCTGCTTGCCCATACTA | 912–894 | 55.6 °C | This study |
| HIV_1270_Rev | CTTCTATTACTTTTACCCATG | 1270–1250 | 45.9 °C | This study |
| HIV_1505_Rev | GTTCCTGCTATRTCACTTCC | 1505–1486 | 51.5 °C | This study |
| HIV_2095_Rev | TCCCTAAAAAATTAGCCTGTC | 2095–2075 | 50.2 °C | This study |
| HIV_3577_Rev | TGATAAATTTGATATGTCCA | 3577–3558 | 43.9 °C | This study |
| HIV_5060_Rev | ACCTGCCATCTGTTTTCCATA | 5060–5040 | 54.2 °C | This study |
| HIV_5537_Rev | ACACTAGGCAAAGGYGG | 5537–5521 | 53.5 °C | This study |
| HIV_6352_Rev | GGTACCCCATAATAGACTGTRACCCACAA | 6352–6324 | 59.9 °C | [25] |
| HIV_6764_Rev | ATCAAGTCTATAAAAAAGTG | 6764–6745 | 42.7 °C | This study |
| HIV_6841_Rev | GGACAAGCCTGTKTAA | 6841–6826 | 47.2 °C | This study |
| HIV_6867_Rev | TGGGAATTGGCTCAAA | 6867–6852 | 48.1 °C | This study |
| HIV_6904_Rev | TTTAGAATCGCAAAACCAGC | 6904–6885 | 51.4 °C | This study |
| HIV_7025_Rev | TTCTGCTAGRCTGCCATT | 7025–7008 | 52.4 °C | This study |
| HIV_7085_Rev | CTGTACTATTATGGTTT | 7085–7069 | 39.1 °C | This study |
| HIV_7351_Rev | AAACTATGTGTTGTAATTTC | 7351–7332 | 43.5 °C | This study |
| HIV_7819_Rev | GCGCCCATAGTGCTTCC | 7819–7803 | 56.7 °C | This study |
| HIV_7845_Rev | CCYGTACCGTCAGCGT | 7845–7830 | 56 °C | This study |
| HIV_8075_Rev | TTTTTACACCGTCTAG | 8075–8059 | 42 °C | This study |
| HIV_8448_Rev | TGTCTTGCTCKCCACCT | 8448–8432 | 42 °C | This study |
| HIV_8528_Rev | GTAGCTGAAGAGGCACAG | 8528-8511 | 54.9 °C | This study |
| HIV_8529_Rev | GGTAGCTGAAGAGGCACAGG | 8529-8510 | 52.6 °C | This study |
| REV14 | ACCATGTTATTTTTCCACATGTTAAA | 6526–6501 | 54.6 °C | [31] |
| REV15 | CTGCCATTTAACAGCAGTTGAGTTGA | 7015–6990 | 57.8 °C | [31] |
| REV 16 | ATGGGAGGGGCATACATTGCT | 7540–7520 | 59 °C | [31] |
| REV 17 | CCTGGAGCTGTTTAATGCCCCAGAC | 7956–7932 | 61.7 °C | [31] |
| REV 18 | GGTGAGTATCCCTGCCTAACTCTAT | 8365–8341 | 57.2 °C | [31] |
| REV 19 | ACTTTTTGACCACTTGCCACCCAT | 8820–8797 | 59.6 °C | [31] |
| Near Full Genome 1 | Near Full Genome 2 | Near Full Genome 3 | Near Full Genome 4 | Half Genome 1a | Half Genome 1b | Half Genome 2 | gagpol | vif/gp120 | env | gp120 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1st Rd Primers | 5’UTRfor1/ 3’UTRrev1 | HIV682for/ HIV9555rev | HIV682for/ 3’UTRrev3 | HIV682for/ 3’UTRrev5 | HIV682for/ Vpu2 rev | Gag3for/ Vpu2 rev | Vif2for/ OFM19 | Gag2for/ pol2 rev | Vif1for/ Gp120out | Vif1/ OFM19 | EnvA/ Gp120out | |
| Sample ID | 2nd Rd Primers | 5’UTRfor2/ 3’UTR rev4 | HIV776for/ HIV9555rev | HIV776for/ 3’UTR rev4 | HIV776for/ 3’UTR rev6 | HIV776for/ Vpu3rev | Gag4for/ Vpu3 rev | Vif3for/ HIV9555rev | Gag3for/ pol3rev | Vif2for/ Gp120in | EnvA/ 02AG-EnvN | EnvB/ Gp120in |
| LB016-1 | positive | − | − | − | positive | positive | positive | − | positive | positive | positive | |
| LB069-1 | positive | − | − | − | − | positive | positive | positive | − | positive | positive | |
| LB082-1 | − | − | − | − | − | positive | positive | positive | positive | positive | positive | |
| LB089-1 | positive | − | positive | − | − | positive | positive | positive | positive | positive | positive | |
| LB095-1 | positive | − | − | − | positive | positive | positive | positive | − | positive | positive | |
| LB104-1 | − | − | − | − | − | positive | positive | − | positive | − | positive | |
| MDC131-1 | − | − | − | − | positive | − | positive | positive | − | − | − | |
| MDC179-2 | − | − | − | − | − | positive | positive | positive | positive | − | − | |
| BDHS24-2 | − | − | − | − | − | positive | positive | positive | positive | positive | positive | |
| BDHS33 | − | − | − | − | − | − | positive | positive | positive | − | positive | |
| NYU119-3 | − | − | − | − | positive | positive | positive | − | positive | − | positive | |
| NYU124-2 | − | − | − | − | positive | positive | positive | − | positive | positive | positive | |
| NYU129-5 | − | positive | − | − | − | positive | positive | positive | − | positive | positive | |
| NYU1122-1 | − | positive | − | positive | positive | positive | positive | − | positive | − | positive | |
| NYU1999 | − | − | − | − | − | positive | positive | positive | − | − | positive | |
| NYU2140-1 | − | − | − | − | positive | − | positive | − | positive | positive | positive | |
| NYU6556-3 | − | − | − | positive | − | positive | positive | positive | positive | positive | − | |
| NYU6541-6 | positive | positive | positive | positive | positive | positive | positive | − | positive | positive | positive | |
| NYU6506-8 | − | − | − | − | − | positive | positive | − | − | − | − | |
| Strain0526 | − | − | − | − | positive | − | positive | − | − | − | − | |
| LB002-1 | − | − | positive | − | NA | NA | NA | NA | NA | NA | NA | |
| LB022-1 | − | − | positive | − | NA | NA | NA | NA | NA | NA | NA | |
| LB006-1 | − | − | − | positive | NA | NA | NA | NA | NA | NA | NA | |
| Miscellaneous HIV-1 genome regions amplified | ||||||||||||
| gag | pol | vifvpu | gp120 | gp41/nef | ||||||||
| 1st Rd primers | Gag2for/ Gag1 rev | Pol1for/ Pol1rev | Vif1for/ Vpu1rev | EnvA/ Gp120out | Gp120in for1/ 3’UTRrev1 | |||||||
| Sample ID | 2nd Rd primers | Gag4for/ Gag3rev | Pol2for/ Pol2rev | Vif2for/ Vpu2rev | EnvB/ Gp120in | Gp120in for2/ 3’UTRrev4 | ||||||
| MDC131-1 | positive | positive | positive | NA | positive | |||||||
| NYU119-3 | positive | positive | positive | positive | positive | |||||||
| NYU124-2 | positive | positive | positive | positive | positive | |||||||
| # | Sample ID | TGS Total Number of HIV Reads | TGS Average Read Length | TGS Number of Long HIV Reads | TGS Genomic Region | SGA Genomic Region |
|---|---|---|---|---|---|---|
| 1 | LB016-1 | 713 | 1731 | 17 (>5000 bp) | Near full genome 1 | NA |
| 2 | LB069-1 | 1701 | 3929 | 488 (>5000 bp) | Near full genome 1 | NA |
| 3 | LB082-1 | 610 | 4232 | 179 (>4000 bp) | Half genome 2 | Half genome 2 |
| 4 | LB089-1 | NA | NA | NA | NA | NA |
| 5 | LB095-1 | NA | NA | NA | NA | Half genome 2 |
| 6 | LB104-1 | 1120 | 3830 | 61 (>4000 bp) | Half genome 2 | NA |
| 7 | MDC131-1 | NA | NA | NA | NA | NA |
| 8 | MDC179-2 | 1956 | 2807 | 504 (>2000 bp) | vif/gp120 | NA |
| 9 | BDHS024-2 | 3463 | 4047 | 799 (>4000 bp) | Half genome 2 | NA |
| 10 | BDHS33 | 3184 | 4122 | 813 (>4000 bp) | Half genome 2 | NA |
| 11 | NYU119-3 | NA | NA | NA | NA | NA |
| 12 | NYU124-2 | 2076 | 2837 | 964 (>2000 bp) | vif/gp120 | NA |
| 13 | NYU129-5 | NA | NA | NA | NA | Half genome 2 |
| 14 | NYU1122-1 | 2648 | 2478 | 752 (>2000 bp) | vif/gp120 | NA |
| 15 | NYU1999-1 | NA | NA | NA | NA | NA |
| 16 | NYU2140-1 | 2108 | 4044 | 448 (>4000 bp) | Half genome 2 | NA |
| 17 | NYU6556-3 | NA | NA | NA | NA | Half genome 2 |
| 18 | NYU6541-6 | 262 | 1415 | 41 (>5000 bp) | Near full genome 1 | Near full genome 1 |
| 19 | NYU6506-8 | NA | NA | NA | NA | NA |
| 20 | Strain0526 | NA | NA | NA | NA | Half genome 2 |
| 21 | LB002-1 | NA | NA | NA | NA | NA |
| 22 | LB022-1 | NA | NA | NA | NA | NA |
| 23 | LB006-1 | NA | NA | NA | NA | NA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banin, A.N.; Tuen, M.; Bimela, J.S.; Tongo, M.; Zappile, P.; Khodadadi-Jamayran, A.; Nanfack, A.J.; Meli, J.; Wang, X.; Mbanya, D.; et al. Development of a Versatile, Near Full Genome Amplification and Sequencing Approach for a Broad Variety of HIV-1 Group M Variants. Viruses 2019, 11, 317. https://doi.org/10.3390/v11040317
Banin AN, Tuen M, Bimela JS, Tongo M, Zappile P, Khodadadi-Jamayran A, Nanfack AJ, Meli J, Wang X, Mbanya D, et al. Development of a Versatile, Near Full Genome Amplification and Sequencing Approach for a Broad Variety of HIV-1 Group M Variants. Viruses. 2019; 11(4):317. https://doi.org/10.3390/v11040317
Chicago/Turabian StyleBanin, Andrew N., Michael Tuen, Jude S. Bimela, Marcel Tongo, Paul Zappile, Alireza Khodadadi-Jamayran, Aubin J. Nanfack, Josephine Meli, Xiaohong Wang, Dora Mbanya, and et al. 2019. "Development of a Versatile, Near Full Genome Amplification and Sequencing Approach for a Broad Variety of HIV-1 Group M Variants" Viruses 11, no. 4: 317. https://doi.org/10.3390/v11040317
APA StyleBanin, A. N., Tuen, M., Bimela, J. S., Tongo, M., Zappile, P., Khodadadi-Jamayran, A., Nanfack, A. J., Meli, J., Wang, X., Mbanya, D., Ngogang, J., Heguy, A., Nyambi, P. N., Fokunang, C., & Duerr, R. (2019). Development of a Versatile, Near Full Genome Amplification and Sequencing Approach for a Broad Variety of HIV-1 Group M Variants. Viruses, 11(4), 317. https://doi.org/10.3390/v11040317