Hepatitis B Virus X Gene Differentially Modulates Subgenotype F1b and F4 Replication

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture
2.2. Viral Variants
2.3. HBx Expression Plasmids
2.4. Transient Transfection
2.5. Transfection Efficiency
2.6. Analysis of Intracellular HBV DNA
2.7. Analysis of Extracellular HBV DNA
2.8. Analysis of Pre-Genomic RNA
2.9. Statistical Analysis
3. Results
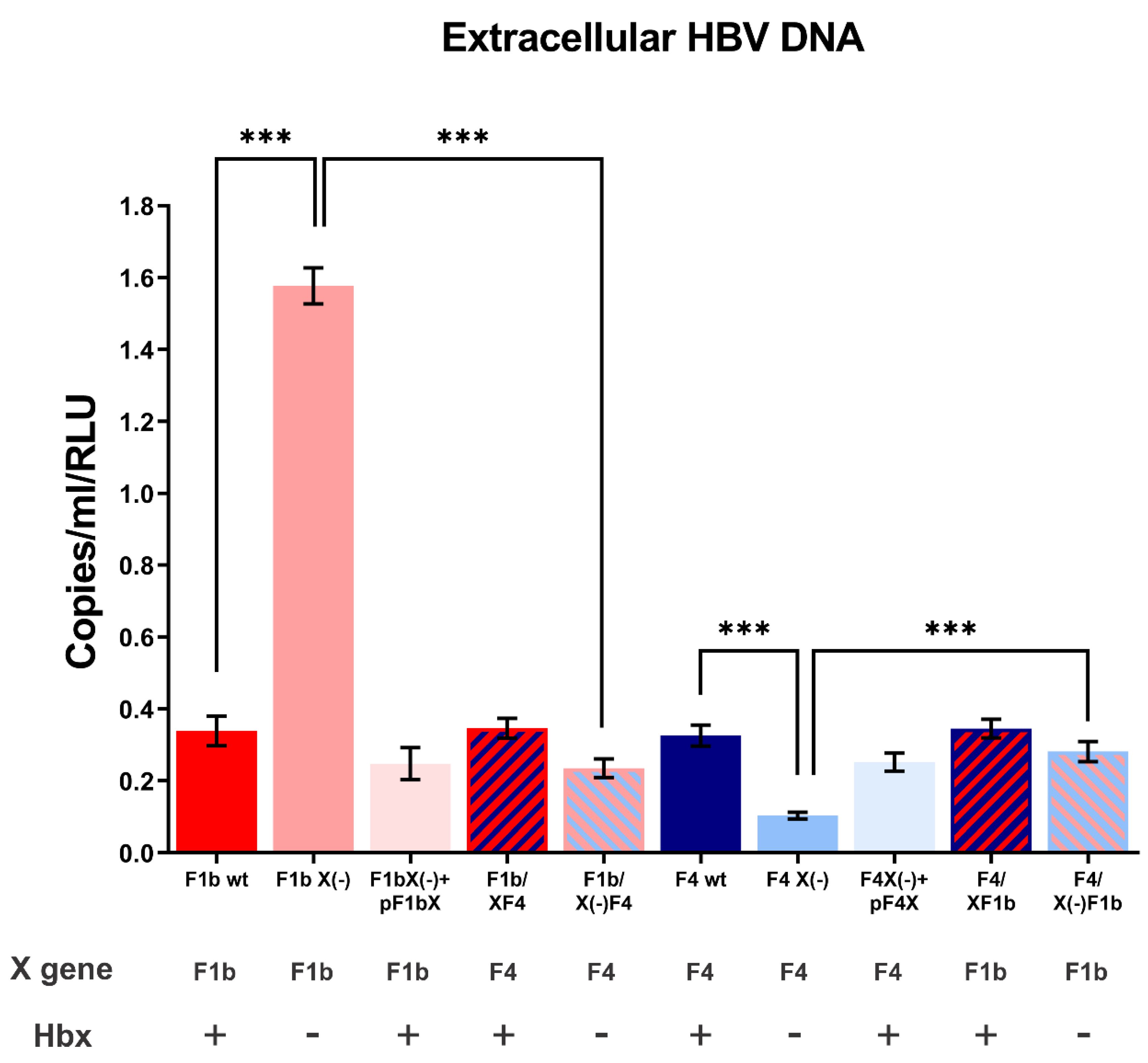
3.1. HBx Modulates Wild Type Levels of sgtF1b and sgtF4 HBV Replication in HuH-7 cells
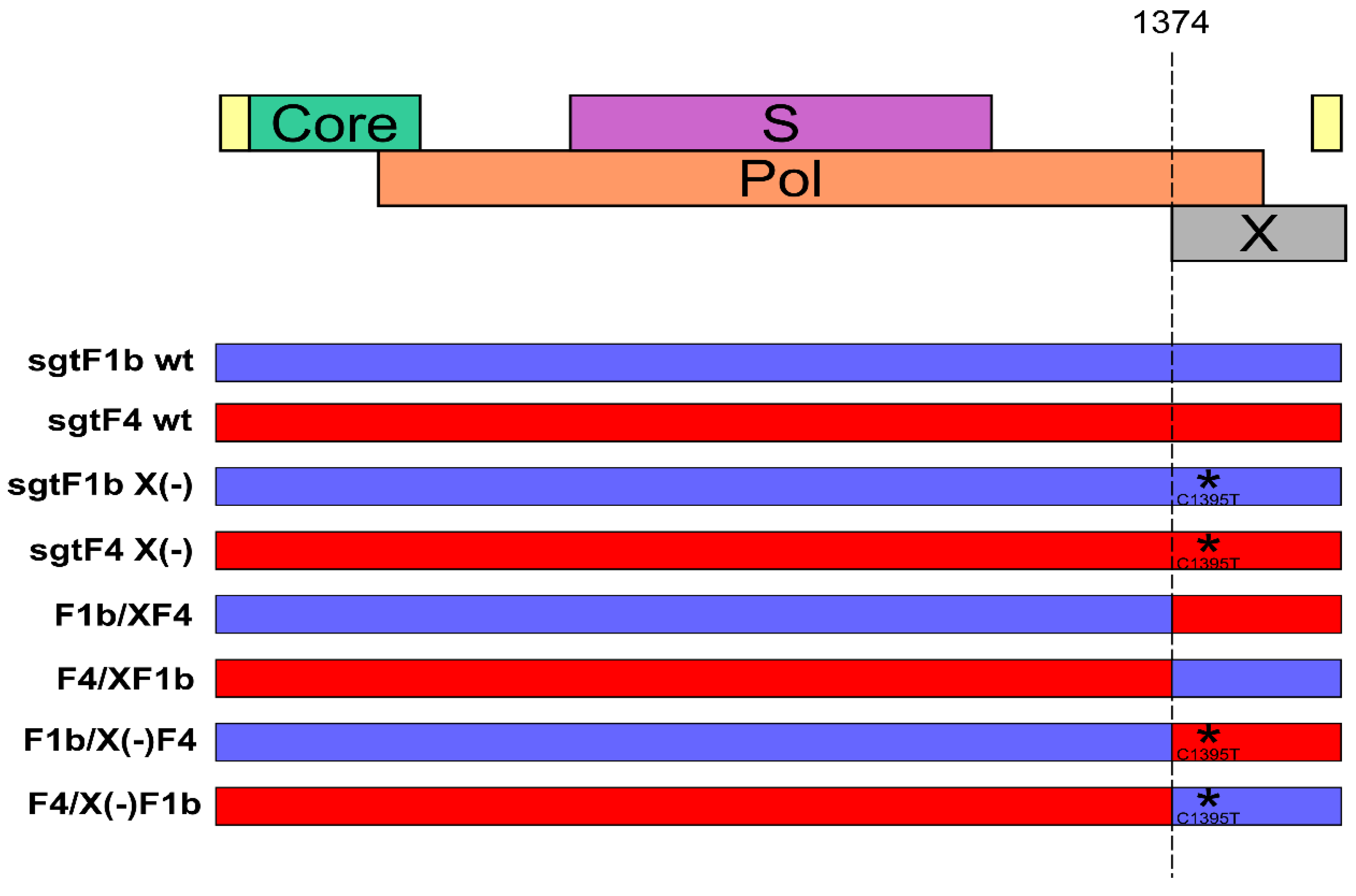
3.2. X Gene Sequence Regulates sgtF1b and sgtF4 HBV Replication
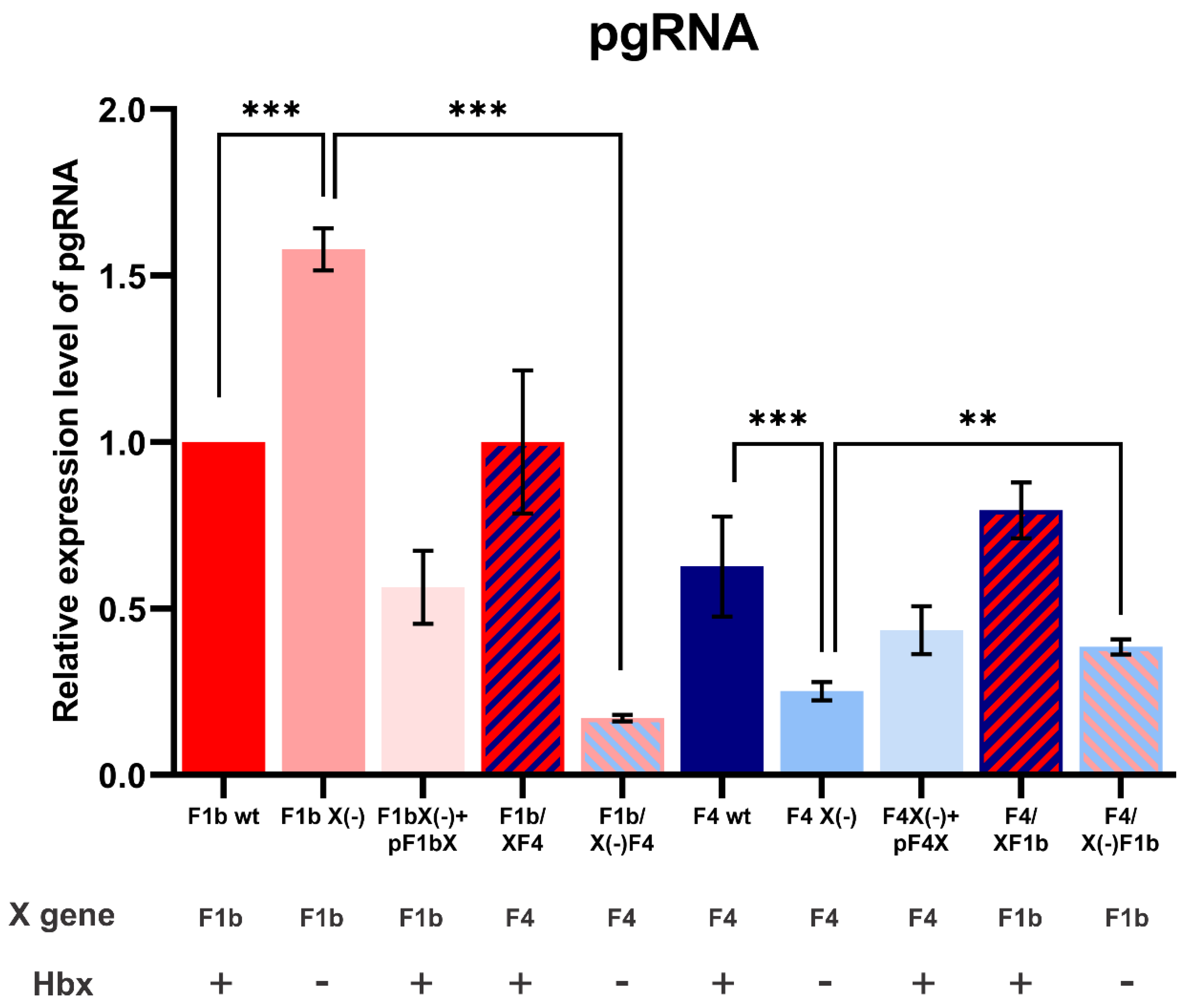
3.3. X Gene Sequence of sgtF1b and sgtF4 Modulates pgRNA Transcription
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Hepatitis B; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Caballero, A.; Tabernero, D.; Buti, M.; Rodriguez-Frias, F. Hepatitis B virus: The challenge of an ancient virus with multiple faces and a remarkable replication strategy. Antivir. Res. 2018, 158, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Elizalde, M.M.; Campos, R.; Barbini, L. X protein variants of the autochthonous Latin American hepatitis B virus F genotype promotes human hepatocyte death by the induction of apoptosis and autophagy. Virus Res. 2017, 242, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Elizalde, M.M.; Sevic, I.; González López Ledesma, M.M.; Campos, R.H.; Barbini, L.; Flichman, D.M. Human hepatocytes apoptosis induced by replication of hepatitis B virus subgenotypes F1b and F4: Role of basal core promoter and preCore mutations. Virology 2018, 513, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Slagle, B.L.; Bouchard, M.J. Hepatitis B virus X and regulation of viral gene expression. Cold Spring Harb. Perspect. Med. 2016, 6, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Delgermaa, L.; Huang, F.; Oishi, N.; Liu, L.; He, F. The Transcriptional Transactivation Function of HBx Protein Is Important for Its Augmentation Role in Hepatitis B Virus Replication. J. Virol. 2005, 79, 5548–5556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, D.Y.; Chen, E.Q.; Huang, F.J.; Leng, X.H.; Cheng, X.; Tang, H. Role and Functional Domain of Hepatitis B Virus X Protein in Regulating HBV Transcription and Replication in Vitro and in Vivo. Viruses 2013, 5, 1261–1271. [Google Scholar] [CrossRef] [Green Version]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [Green Version]
- Pollicino, T.; Belloni, L.; Raffa, G.; Pediconi, N.; Squadrito, G.; Raimondo, G.; Levrero, M. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 2006, 130, 823–837. [Google Scholar] [CrossRef]
- Luo, L.; Chen, S.; Gong, Q.; Luo, N.; Lei, Y.; Guo, J.; He, S. Hepatitis B virus X protein modulates remodelling of minichromosomes related to hepatitis B virus replication in HepG2 cells. Int. J. Mol. Med. 2013, 31, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Kramvis, A. Genotypes and genetic variability of hepatitis B virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Zhang, Z.; Ling, C.; Zheng, W.; Zhu, C.; Carr, M.J.; Higgins, D.G. Hepatitis B virus subgenotyping: History, effects of recombination, misclassifications, and corrections. Infect. Genet. Evol. 2013, 16, 355–361. [Google Scholar] [CrossRef]
- Lin, C.L.; Kao, J.H. The clinical implications of hepatitis B virus genotype: Recent advances. J. Gastroenterol. Hepatol. 2011, 26, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Kao, J.H. Global perspective on the natural history of chronic hepatitis B: Role of hepatitis B virus genotypes A to J. Semin. Liver Dis. 2013, 33, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Mojsiejczuk, L.; Elizalde, M.M.; López, G.; Figueredo, D.; Marquez, N.; Campos, R.H.; Flichman, D. Molecular epidemiology of hepatitis B virus in Paraguay. Infect. Genet. Evol. 2019, 71, 91–97. [Google Scholar] [CrossRef]
- Pezzano, S.C.; Torres, C.; Fainboim, H.A.; Bouzas, M.B.; Schroder, T.; Giuliano, S.F.; Paz, S.; Alvarez, E.; Campos, R.H.; Mbayed, V.A. Hepatitis B virus in Buenos Aires, Argentina: Genotypes, virological characteristics and clinical outcomes. Clin. Microbiol. Infect. 2011, 17, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Ledesma, M.M.G.L.; Mojsiejczuk, L.N.; Rodrigo, B.; Sevic, I.; Mammana, L.; Galdame, O.; Gadano, A.; Fainboim, H.; Campos, R.; Flichman, D. Hepatitis B virus genotype distribution and genotype-specific BCP/precore substitutions in acute and chronic infections in Argentina. PLoS ONE 2015, 10, 1–16. [Google Scholar]
- Barbini, L.; Elizalde, M.; Torres, C.; Campos, R. Molecular epidemiology and genetic diversity of hepatitis B virus in Mar del Plata city, Argentina. Infect. Genet. Evol. 2013, 19, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Sevic, I.; González López Ledesma, M.M.; Flichman, D.M.; Campos, H.R. HBV DNA genome co-transfection procedure for the evaluation of relative fitness. PLoS ONE 2017, 12, 1–12. [Google Scholar] [CrossRef]
- Sevic, I.; Elizalde, M.M.; González López Ledesma, M.M.; Flichman, D.M.; Campos, R.H. Analysis of fitness differences of hepatitis B virus genotypes D and F using a cotransfection assay. Arch. Virol. 2018, 164, 447–455. [Google Scholar] [CrossRef]
- Ching, L.K.; Gounder, P.P.; Bulkow, L.; Spradling, P.R.; Bruce, M.G.; Negus, S.; Snowball, M.; McMahon, B.J. Incidence of hepatocellular carcinoma according to hepatitis B virus genotype in Alaska Native people. Liver Int. 2016, 36, 1507–1515. [Google Scholar] [PubMed] [Green Version]
- Gounder, P.P.; Bulkow, L.R.; Snowball, M.; Negus, S.; Spradling, P.R.; McMahon, B.J. Hepatocellular Carcinoma Risk in Alaska Native Children and Young Adults with Hepatitis B Virus: Retrospective Cohort Analysis. J. Pediatr. 2016, 178, 206–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pineau, P.; Ruiz, E.; Deharo, E.; Bertani, S. On hepatocellular carcinoma in South America and early-age onset of the disease. Clin. Res. Hepatol. Gastroenterol. 2018, 24, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bertani, S.; Pineau, P.; Loli, S.; Moura, J.; Zimic, M.; Deharo, E.; Ruiz, E. An Atypical Age-Specific Pattern of Hepatocellular Carcinoma in Peru: A Threat for Andean Populations. PLoS ONE 2013, 8, e67756. [Google Scholar] [CrossRef]
- Nakabayashi, H.; Miyano, K.; Sato, J.; Yamane, T.; Taketa, K. Growth of human hepatoma cell lines with differentiated functions in chemically defined medium. Cancer Res. 1982, 42, 3858–3863. [Google Scholar] [PubMed]
- Günther, S.; Li, B.C.; Miska, S.; Krüger, D.H.; Meisel, H.; Will, H. A novel method for efficient amplification of whole hepatitis B virus genomes permits rapid functional analysis and reveals deletion mutants in immunosuppressed patients. J. Virol. 1995, 69, 5437–5444. [Google Scholar] [Green Version]
- Nassal, M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol. 1992, 66, 4107–4116. [Google Scholar]
- Castro, E.F.; Cavallaro, L.V.; Campos, R.H.; Finkielsztein, L.M.; Fabian, L.E.; Gagey, D.; Caputto, M.E.; Moltrasio, G.Y.; Moglioni, A.G. Inhibition of Bovine Viral Diarrhea Virus RNA Synthesis by Thiosemicarbazone Derived from 5,6-Dimethoxy-1-Indanone. J. Virol. 2011, 85, 5436–5445. [Google Scholar] [CrossRef] [Green Version]
- Keasler, V.V.; Hodgson, A.J.; Madden, C.R.; Slagle, B.L. Enhancement of Hepatitis B Virus Replication by the Regulatory X Protein In Vitro and In Vivo. J. Virol. 2006, 81, 2656–2662. [Google Scholar] [CrossRef]
- Yuh, C.H.; Ting, L.P. Differentiated liver cell specificity of the second enhancer of hepatitis B virus. J. Virol. 1993, 67, 142–149. [Google Scholar] [Green Version]
- Chang, H.; Choul, C.K.; Chang, C.; Su, T.S.; Hul, C.; Mitsuaki, Y.; Ting, L. The enhancer sequence of human hepatitis B virus can enhance the activity of its surface gene promoter. Nucleic Acids Res. 1987, 15, 2261–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quarleri, J. Core promoter: A critical region where the hepatitis B virus makes decisions. World J. Gastroenterol. 2014, 20, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Quasdorff, M.; Protzer, U. Control of hepatitis B virus at the level of transcription. J. Viral Hepat. 2010, 17, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Moolla, N.; Kew, M.; Arbuthnot, P. Regulatory elements of hepatitis B virus transcription. J. Viral Hepat. 2002, 9, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhou, X.; Jia, H.; Chen, C.; Zhao, W.; Zhang, J.; Tong, S. Stronger enhancer II/core promoter activities of hepatitis B virus isolates of B2 subgenotype than those of C2 subgenotype. Sci. Rep. 2016, 6, 30374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajoriya, N.; Combet, C.; Zoulim, F.; Janssen, H.L.A. How viral genetic variants and genotypes influence disease and treatment outcome of chronic hepatitis B. Time for an individualised approach? J. Hepatol. 2017, 67, 1281–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, B.J. The influence of hepatitis B virus genotype and subgenotype on the natural history of chronic hepatitis B. Hepatol. Int. 2009, 3, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Tang, X.; Garcia, T.; Hussain, M.; Zhang, J.; Lok, A.; Wands, J.; Li, J.; Tong, S. Hepatitis B Virus Genotype C Isolates with Wild-Type Core Promoter Sequence Replicate Less Efficiently than Genotype B Isolates but Possess Higher Virion Secretion Capacity. J. Virol. 2011, 85, 10167–10177. [Google Scholar] [CrossRef] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elizalde, M.M.; Speroni, M.; Campos, R.H.; Flichman, D.M. Hepatitis B Virus X Gene Differentially Modulates Subgenotype F1b and F4 Replication. Viruses 2019, 11, 655. https://doi.org/10.3390/v11070655
Elizalde MM, Speroni M, Campos RH, Flichman DM. Hepatitis B Virus X Gene Differentially Modulates Subgenotype F1b and F4 Replication. Viruses. 2019; 11(7):655. https://doi.org/10.3390/v11070655
Chicago/Turabian StyleElizalde, María Mercedes, Micaela Speroni, Rodolfo Héctor Campos, and Diego Martín Flichman. 2019. "Hepatitis B Virus X Gene Differentially Modulates Subgenotype F1b and F4 Replication" Viruses 11, no. 7: 655. https://doi.org/10.3390/v11070655
APA StyleElizalde, M. M., Speroni, M., Campos, R. H., & Flichman, D. M. (2019). Hepatitis B Virus X Gene Differentially Modulates Subgenotype F1b and F4 Replication. Viruses, 11(7), 655. https://doi.org/10.3390/v11070655