The Ecology and Phylogeny of Hosts Drive the Enzootic Infection Cycles of Hantaviruses

Abstract
:1. Introduction
2. Materials and Methods
2.1. Site Data Collection
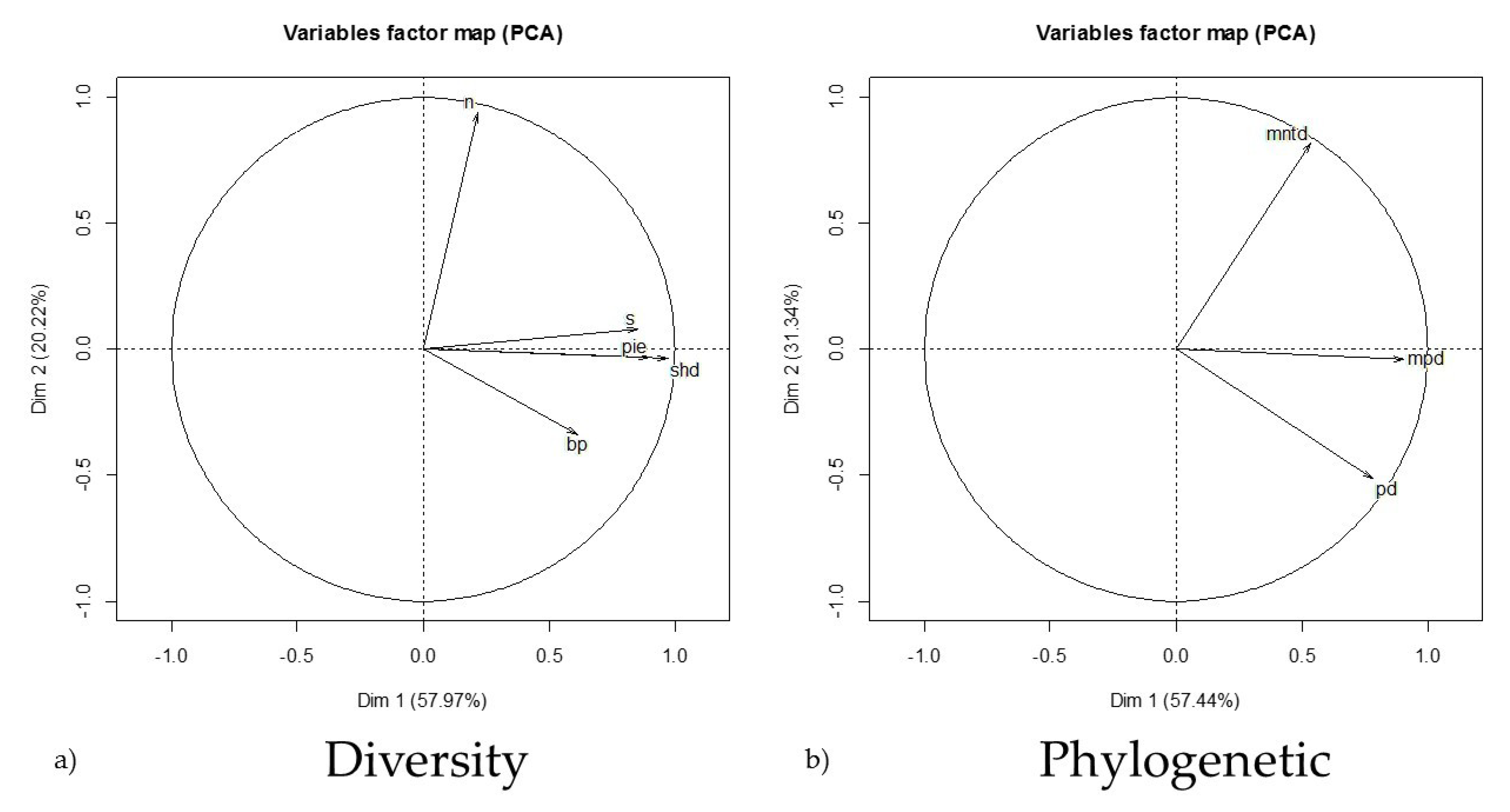
2.2. Response and Predictor Variables
2.3. Statistical Analysis
3. Results
3.1. Sites and Variables
3.2. Model Selection
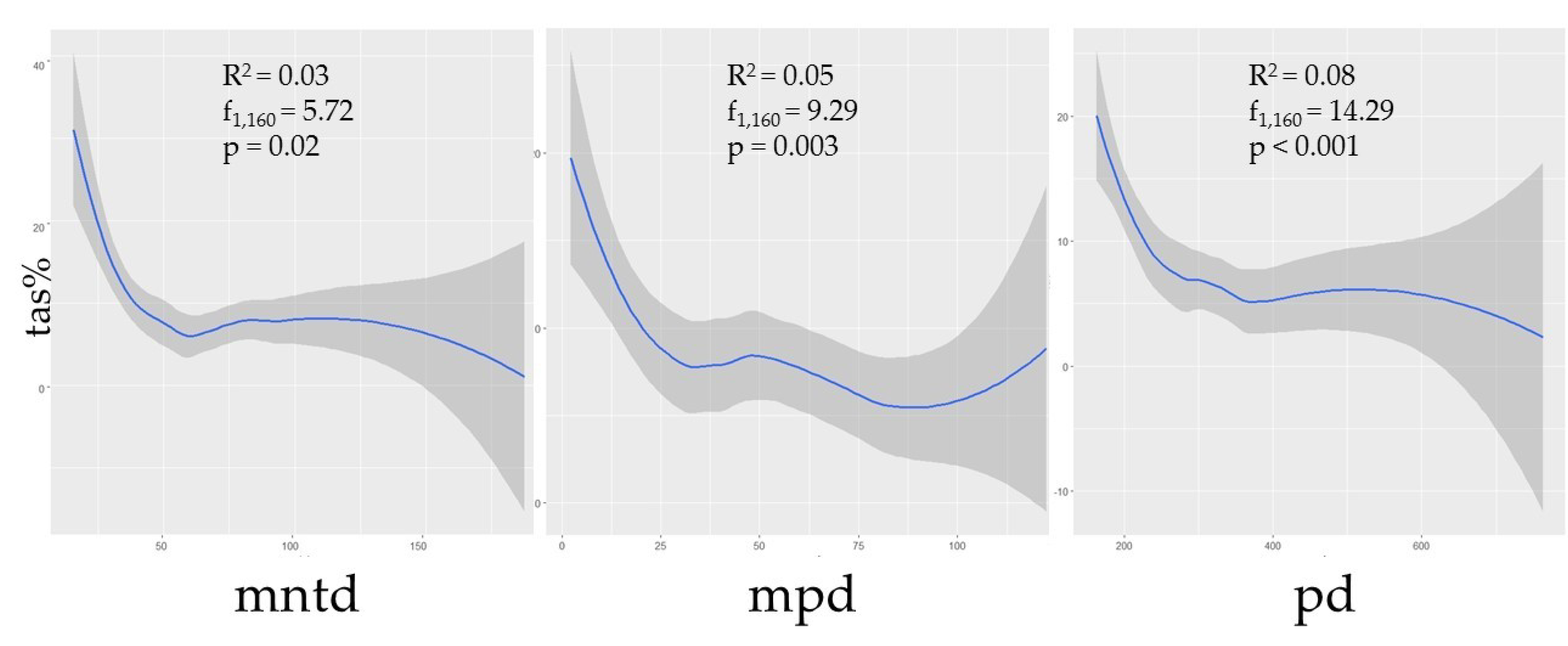
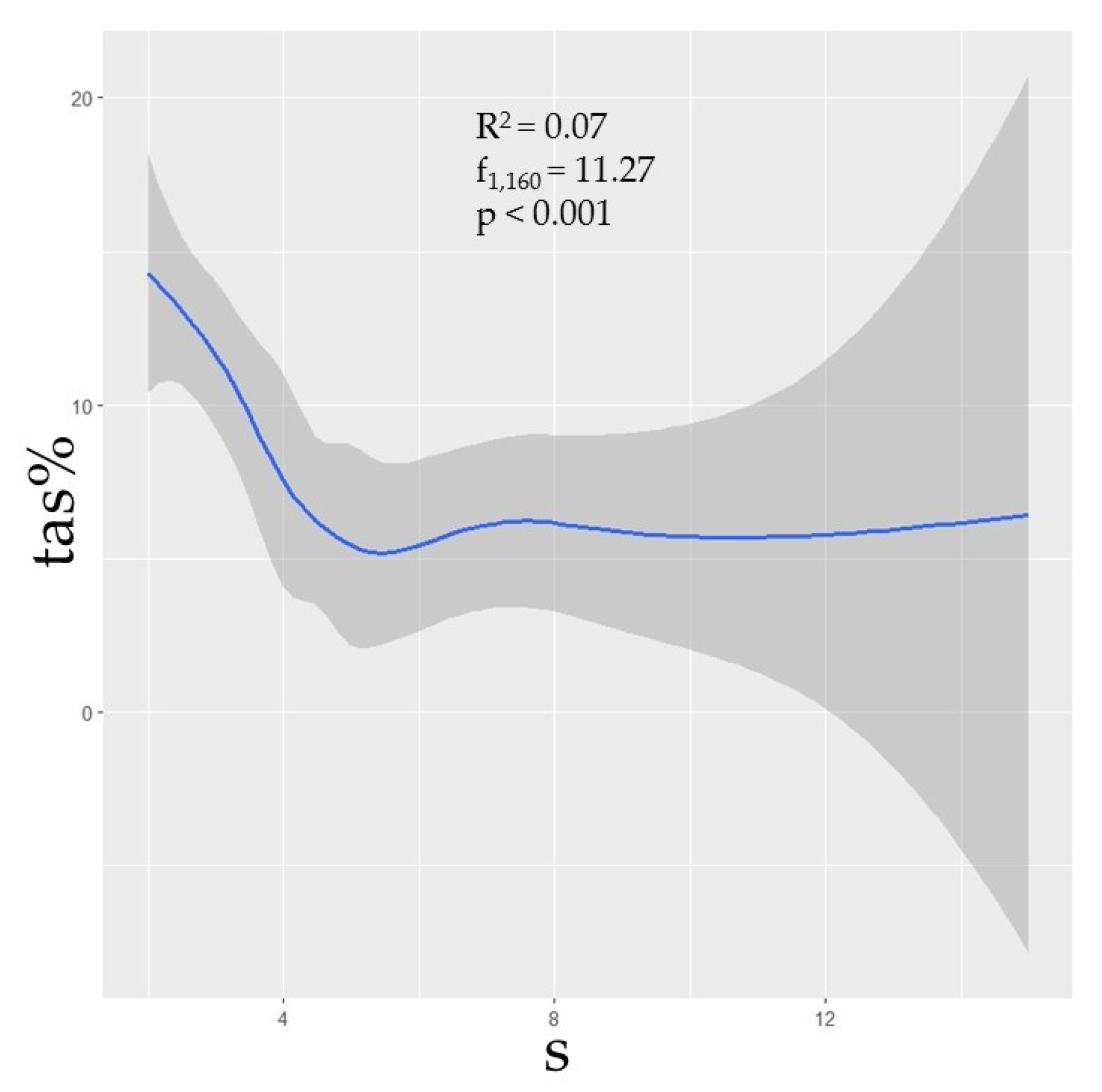
3.3. Species Diversity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abudurexiti, A.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Avšič-Županc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, É.; Blair, C.D.; et al. Taxonomy of the order Bunyavirales: Update 2019. Arch. Virol. 2019, 164, 1949–1965. [Google Scholar] [CrossRef] [PubMed]
- Chandy, S.; Abraham, S.; Sridharan, G.; Abraham, P.; Sridharan, G. Hantaviruses: An emerging public health threat in India? A review. (Special issue. Emerging and re-emerging infections in India). J. Biosci. 2008, 33, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Knust, B.; Rollin, P.E. Twenty-year summary of surveillance for human hantavirus infections, United States. Emerg. Infect. Dis. 2013, 19, 1934–1937. [Google Scholar] [CrossRef] [PubMed]
- Bi, Z.; Formenty, P.B.H.; Roth, C.E. Hantavirus infection: A review and global update. J. Infect. Dev. Ctries. 2008, 2, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Milholland, M.T.; Castro-Arellano, I.; Suzán, G.; Garcia-Peña, G.E.; Lee, T.E.; Rohde, R.E.; Alonso Aguirre, A.; Mills, J.N. Global diversity and distribution of hantaviruses and their hosts. Ecohealth 2018, 15, 163–208. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, C.B.; Figueiredo, L.T.M.; Vapalahti, O. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 2010, 23, 412–441. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, J.; Thomazelli, L.M.; Henriques, D.A.; Lautenschalager, D.; Ometto, T.; Dutra, L.M.; Aires, C.C.; Favorito, S.; Durigon, E.L. Detection of hantavirus in bats from remaining rain forest in São Paulo, Brazil. BMC Res. Notes 2015, 5, 690. [Google Scholar] [CrossRef]
- Guo, W.P.; Lin, X.D.; Wang, W.; Tian, J.H.; Cong, M.L.; Zhang, H.L.; Wang, M.R.; Zhou, R.H.; Wang, J.B.; Li, M.H.; et al. Phylogeny and Origins of Hantaviruses Harbored by Bats, Insectivores, and Rodents. PLoS Pathog. 2013, 9, e1003159. [Google Scholar] [CrossRef] [PubMed]
- Torres-Pérez, F.; Wilson, L.; Collinge, S.K.; Harmon, H.; Ray, C.; Medina, R.A.; Hjelle, B. Sin nombre virus infection in field workers, Colorado, USA. Emerg. Infect. Dis. 2010, 16, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Arellano, E.; Castro-Arellano, I.; Suzán, G.; González-Cózatl, F.X.; Jiménez, R.M. Antibody Seroprevalence to Hantaviruses in Rodents from Reserva De La Biosfera Sierra De Huautla, Morelos. West. North Am. Nat. 2012, 72, 105–109. [Google Scholar] [CrossRef]
- Song, J.W.; Hae, J.K.; Song, K.J.; Truong, T.T.; Bennett, S.N.; Arai, S.; Truong, N.U.; Yanagihara, R. Newfound hantavirus in Chinese mole shrew, Vietnam. Emerg. Infect. Dis. 2007, 13, 1784–1787. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Ohdachi, S.D.; Asakawa, M.; Kang, H.J.; Mocz, G.; Arikawa, J.; Okabe, N.; Yanagihara, R. Molecular phylogeny of a newfound hantavirus in the Japanese shrew mole (Urotrichus talpoides). Proc. Natl. Acad. Sci. USA 2008, 105, 16296–16301. [Google Scholar] [CrossRef]
- Kang, H.J.; Kadjo, B.; Dubey, S.; Jacquet, F.; Yanagihara, R. Molecular evolution of Azagny virus, a newfound hantavirus harbored by the West African pygmy shrew (Crocidura obscurior) in Côte d’Ivoire. Virol. J. 2011, 8, 373. [Google Scholar] [CrossRef]
- Kang, H.J.; Bennett, S.N.; Hope, A.G.; Cook, J.A.; Yanagihara, R. Shared Ancestry between a Newfound Mole-Borne Hantavirus and Hantaviruses Harbored by Cricetid Rodents. J. Virol. 2011, 85, 7496–7503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, S.; Nguyen, S.T.; Boldgiv, B.; Fukui, D.; Araki, K.; Dang, C.N.; Ohdachi, S.D.; Nguyen, N.X.; Pham, T.D.; Boldbaatar, B.; et al. Novel Bat-borne Hantavirus, Vietnam. Emerg. Infect. Dis. 2013, 19, 1159–1161. [Google Scholar] [CrossRef]
- Sumibcay, L.; Kadjo, B.; Gu, S.H.; Kang, H.J.; Lim, B.K.; Cook, J.A.; Song, J.W.; Yanagihara, R. Divergent lineage of a novel hantavirus in the banana pipistrelle (Neoromicia nanus) in Côte d’Ivoire. Virol. J. 2012, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Hinson, E.R.; Shone, S.M.; Zink, M.C.; Glass, G.E.; Klien, S.L. Wounding: The primary mode of Seoul virus transmission among male Norway rats. Am. J. Trop. Med. Hyg. 2004, 70, 310–317. [Google Scholar] [CrossRef]
- McIntyre, N.E.; Chu, Y.K.; Owen, R.D.; Abuzeineh, A.; de La Sancha, N.; Dick, C.W.; Holsomback, T.; Nisbett, R.A.; Jonsson, C. A longitudinal study of Bayou virus, hosts, and habitat. Am. J. Trop. Med. Hyg. 2005, 73, 1043–1049. [Google Scholar] [CrossRef]
- Kariwa, H.; Yoshida, H.; Sánchez-Hernández, C.; de Lourdes Romero-Almaraz, M.; Almazán-Catalán, J.A.; Ramos, C.; Miyashita, D.; Seto, T.; Takano, A.; Totani, M.; et al. Genetic diversity of hantaviruses in Mexico: Identification of three novel hantaviruses from Neotominae rodents. Virus Res. 2012, 163, 486–494. [Google Scholar] [CrossRef]
- Schlegel, M.; Jacob, J.; Krüger, D.H.; Rang, A.; Ulrich, R.G. Hantavirus Emergence in Rodents, Insectivores and Bats: What Comes Next? Elsevier: Amsterdam, Netherland, 2013; ISBN 9780124051911. [Google Scholar]
- Mills, J.N.; Ksiazek, T.G.; Ellis, B.A.; Rollin, P.E.; Nichol, S.T.; Yates, T.L.; Gannon, W.L.; Levy, C.E.; Engelthaler, D.M.; Davis, T.; et al. Patterns of association with host and habitat: Antibody reactive with Sin Nombre virus in small mammals in the major biotic communities of the southwestern United States. Am. J. Trop. Med. Hyg. 1997, 56, 273–284. [Google Scholar] [CrossRef]
- Schmaljohn, C.; Hjelle, B. Hantaviruses: A Global Disease Problem. Emerg. Infect. Dis. 1997, 3, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, K.L.; Rollin, P.E.; Shieh, W.J.; Zaki, S.; Greer, P.W.; Peters, C.J. Transmission of Black Creek Canal virus between cotton rats. J. Med. Virol. 2000, 60, 70–76. [Google Scholar] [CrossRef]
- Schountz, T.; Prescott, J. Hantavirus immunology of rodent reservoirs: Current status and future directions. Viruses 2014, 6, 1317–1335. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Pei, K.J.C.; Yang, C.M.; Kuo, M.D.; Wong, S.T.; Kuo, S.C.; Lin, F.G. A possible case of hantavirus infection in a Borneo orangutan and its conservation implication. J. Med. Primatol. 2011, 40, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Lundkvist, Å.; Verner-Carlsson, J.; Plyusnina, A.; Forslund, L.; Feinstein, R.; Plyusnin, A. Pet rat harbouring Seoul hantavirus in Sweden, June 2013. Eurosurveillance 2013, 18, 1–4. [Google Scholar] [CrossRef]
- Taori, S.K.; Jameson, L.J.; Campbell, A.; Drew, P.J.; McCarthy, N.D.; Hart, J.; Osborne, J.C.; Sudhanva, M.; Brooks, T.J. UK hantavirus, renal failure, and pet rats. Lancet 2013, 381, 1070. [Google Scholar] [CrossRef]
- Childs, J.E.; Ksiazek, T.G.; Spiropoulou, C.F.; Krebs, J.W.; Morzunov, S.; Maupin, G.O.; Gage, K.L.; Rollin, P.E.; Sarisky, J.; Enscore, R.E.; et al. Serologic and genetic identification of Peromyscus maniculatus as the primary rodent reservoir for a new hantavirus in the southwestern United States. J. Infect. Dis. 1994, 169, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Smith, J.O.; George, D.; Pepkin, K.M.; Pitzer, V.E.; Pulliam, J.R.; Dobson, A.P.; Hudson, J.P.; Grenfell, B.T. Epidemic dynamics at the human-animal interface. Science 2009, 326, 1362–1367. [Google Scholar] [CrossRef]
- Tian, H.; Yu, P.; Bjørnstad, O.N.; Cazelles, B.; Yang, J.; Tan, H.; Huang, S.; Cui, Y.; Dong, L.; Ma, C.; et al. Anthropogenically driven environmental changes shift the ecological dynamics of hemorrhagic fever with renal syndrome. PLoS Pathog. 2017, 13, 1–19. [Google Scholar] [CrossRef]
- Mills, J.N.; Amman, B.R.; Glass, G.E. Ecology of Hantaviruses and their hosts in North America. Vector-Borne Zoonotic Dis. 2009, 10, 563–574. [Google Scholar] [CrossRef]
- Halliday, J.; Daborn, C.; Auty, H.; Mtema, Z.; Lembo, T.; Bronsvoort, B.M.deC.; Handel, I.; Knobel, D.; Hampson, K.; Cleaveland, S. Bringing together emerging and endemic zoonoses surveillance: Shared challenges and a common solution. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2872–2880. [Google Scholar] [CrossRef] [PubMed]
- Polop, F.J.; Provensal, M.C.; Pini, N.; Levis, S.C.; Priotto, J.W.; Enría, D.; Calderón, G.E.; Costa, F.; Polop, J.J. Temporal and spatial host abundance and prevalence of Andes hantavirus in Southern Argentina. Ecohealth 2010, 7, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Calisher, C.H.; Root, J.J.; Mills, J.N.; Beaty, B.J. Assessment of ecologic and biologic factors leading to hantavirus pulmonary syndrome, Colorado, USA. Croat. Med. J. 2002, 43, 330–337. [Google Scholar]
- Allen, L.J.S.; McCormack, R.K.; Jonsson, C.B. Mathematical models for hantavirus infection in rodents. Bull. Math. Biol. 2006, 68, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.T.J.; Preston, D.L.; Hoverman, J.T.; Lafonte, B.E. Host and parasite diversity jointly control disease risk in complex communities. Proc. Natl. Acad. Sci. USA 2013, 110, 16916–16921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostfeld, R.S.; Keesing, F. Biodiversity and disease risk: The case of Lyme disease. Conserv. Biol. 2000, 14, 722–728. [Google Scholar] [CrossRef]
- Clay, C.A.; Lehmer, E.M.; Jeor, S.S.; Dearing, M.D. Testing mechanisms of the dilution effect: Deer mice encounter rates, Sin Nombre virus prevalence and species diversity. Ecohealth 2009, 6, 250–259. [Google Scholar] [CrossRef]
- Zargar, U.R.; Chishti, M.Z.; Ahmad, F.; Rather, M.I. Does alteration in biodiversity really affect disease outcome?—A debate is brewing. Saudi J. Biol. Sci. 2015, 22, 14–18. [Google Scholar] [CrossRef]
- Luis, A.D.; Kuenzi, A.J.; Mills, J.N. Species diversity concurrently dilutes and amplifies transmission in a zoonotic host–pathogen system through competing mechanisms. Proc. Natl. Acad. Sci. USA 2018, 115, 7979–7984. [Google Scholar] [CrossRef]
- Keesing, F.; Holt, R.D.; Ostfeld, R.S. Effects of species diversity on disease risk. Ecol. Lett. 2006, 9, 485–498. [Google Scholar] [CrossRef]
- Glass, G.E.; Yates, T.L.; Fine, J.B.; Shields, T.M.; Kendall, J.B.; Hope, A.G.; Parmenter, C.A.; Peters, C.J.; Ksiazek, T.G.; Li, C.-S.; et al. Satellite imagery characterizes local animal reservoir populations of Sin Nombre virus in the southwestern United States. Proc. Natl. Acad. Sci. USA 2002, 99, 16817–16822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeimes, C.B.; Olsson, G.E.; Ahlm, C.; Vanwambeke, S.O. Modelling zoonotic diseases in humans: Comparison of methods for hantavirus in Sweden. Int. J. Health Geogr. 2012, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.A.; Preston, N.; Allen, T.; Zambrana-Torrelio, C.; Hosseini, P.R.; Daszak, P. Global biogeography of human infectious diseases. Proc. Natl. Acad. Sci. USA 2015, 112, 12746–12751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordes, F.; Caron, A.; Blasdell, K.; de Garine-Wichatitsky, M.; Morand, S. Forecasting potential emergence of zoonotic diseases in south-east Asia: Network analysis identifies key rodent hosts. J. Appl. Ecol. 2017, 54, 691–700. [Google Scholar] [CrossRef]
- Sauvage, F.; Langlais, M.; Yoccoz, N.G.; Pontier, D.; Yoccozt, N.G. Modeling hantavirus in fluctuating populations of bank voles: The role of indirect transmission on virus persistence. J. Animal Ecol. 2003, 72, 1–13. [Google Scholar] [CrossRef]
- Adler, F.R.; Pearce-Duvet, J.M.C.; Dearing, M.D. How host population dynamics translate into time-lagged prevalence: An Investigation of Sin Nombre virus in deer mice. Bull. Math. Biol. 2008, 70, 236–252. [Google Scholar] [CrossRef] [PubMed]
- Milholland, M.T.; Castro-Arellano, I.; Arellano, E.; Nava-García, E.; Rangel-Altamirano, G.; Gonzalez-Cozatl, F.X.; Suzán, G.; Schountz, T.; González-Padrón, S.; Vigueras, A.; et al. Species identity supersedes the dilution effect concerning hantavirus prevalence at sites across Texas and México. ILAR J. 2017, 58, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.L.; Milholland, M.T.; Schountz, T.; Castro-Arellano, I.; Mali, I. Detection of New World hantavirus antibodies in rodents of eastern New Mexico. J. Wildl. Manag. 2019. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric Estimation of the Number of Classes in a Population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Colwell, R.K.; Elsensohn, J.E. EstimateS turns 20: Statistical estimation of species richness and shared species from samples, with non-parametric extrapolation. Ecography 2014, 37, 609–613. [Google Scholar] [CrossRef]
- EcoSim: Null models software for ecology. Available online: http://www.uvm.edu/~ngotelli/EcoSim/EcoSim.html (accessed on 15 August 2013).
- Magurran, A.E. Measuring Biological Diversity; Blackwell Publishing: Malden, MA, USA, 2004; pp. 19–215. [Google Scholar]
- Hurlbert, S.H. The nonconcept of species diversity: A critique and alternative parameters. Ecology 1971, 52, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Adler, F.R.; Clay, C.A.; Lehmer, E.M. The role of heterogeneity in the persistence and prevalence of Sin Nombre virus in deer mice. Am. Nat. 2008, 172, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Gómez, J.M.; Bosch, J.; Perfectti, F.; Fernández, J.; Abdelaziz, M. Pollinator diversity affects plant reproduction and recruitment: The tradeoffs of generalization. Oecologia 2007, 153, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.O.; Ackerly, D.D.; McPeek, M.A.; Donoghue, M.J. Phylogenies and community ecology. Annu. Rev. Ecol. Syst. 2002, 33, 475–505. [Google Scholar] [CrossRef]
- Webb, C.O.; Ackerly, D.D.; Kembel, S.W. Phylocom: Software for the analysis of phylogenetic community structure and trait evolution. Bioinformatics 2008, 24, 2098–2100. [Google Scholar] [CrossRef]
- Ives, A.R.; Helmus, M.R. Phylogenetic Metrics of Community Similarity. Am. Nat. 2010, 176, E128–E142. [Google Scholar] [CrossRef] [Green Version]
- Clarke, K.R.; Warwick, R.M. A taxonomic distinctness index and its statistical properties. J. Appl. Ecol. 1998, 35, 523–531. [Google Scholar] [CrossRef]
- Barker, G.M. Phylogenetic diversity: A quantitative framework for measurement of priority and achievement in biodiversity conservation. Biol. J. Linn. Soc. Lond. 2002, 76, 165–194. [Google Scholar] [CrossRef]
- Helmus, M.R.; Bland, T.J.; Williams, C.K.; Ives, A.R. Phylogenetic measure of biodiversity. Am. Nat. 2007, 169, 68–83. [Google Scholar] [CrossRef]
- Bininda-Emonds, O.R.P.; Cardillo, M.; Jones, K.E.; MacPhee, R.D.E.; Beck, R.M.D.; Grenyer, R.; Price, S.A.; Vos, R.A.; Gittleman, J.L.; Purvis, A. The delayed rise of present-day mammals. Nature 2007, 446, 507–512. [Google Scholar] [CrossRef]
- Lumley, T. Package “leaps”: Regression Subset Selection, vs. 3.0.; Chapman & Hall: London, UK, 2017. [Google Scholar]
- Husson, F.; Josse, J.; Le, S.; Maintainer, J.M. Package “FactoMineR” Multivariate Exploratory Data Analysis and Data Mining; Chapman & Hall: London, UK, 2017. [Google Scholar]
- Miller, S. Species-specific traits versus site-specific properties as factors influencing the abundances of species. Masters Thesis, Texas State University, San Marcos, TX, USA, 2014. [Google Scholar]
- Scaled PCA: A new approach to dimension reduction. Available online: https://ssrn.com/abstract=3358911 or http://dx.doi.org/10.2139/ssrn.3358911 (accessed on 29 December 2014).
- Akaike, H. Information theory and an extension of the maximum likelihood principle. In International Symposium on Information Theory; Petrov, B.N., Csaaki, F., Eds.; Akadeemai Kiadi: Budapest, Hungary, 1973. [Google Scholar]
- “MuMIn: Multi-model inference”. R package version 1.9.13. Available online: https://cran.r-project.org/web/packages/MuMIn/ (accessed on 8 April 2015).
- Civitello, D.J.; Cohen, J.; Fatima, H.; Halstead, N.T.; Liriano, J.; McMahon, T.A.; Ortega, C.N.; Sauer, E.L.; Sehgal, T.; Young, S.; et al. Biodiversity inhibits parasites: Broad evidence for the dilution effect. Proc. Natl. Acad. Sci. USA 2015, 112, 8667–8671. [Google Scholar] [CrossRef] [Green Version]
- Salkeld, D.J.; Padgett, K.A.; Jones, J.H. A meta-analysis suggesting that the relationship between biodiversity and risk of zoonotic pathogen transmission is idiosyncratic. Ecol. Lett. 2013, 16, 679–686. [Google Scholar] [CrossRef]
- Johnson, P.T.J.; Ostfeld, R.S.; Keesing, F. Frontiers in research on biodiversity and disease. Ecol. Lett. 2015, 18, 1119–1133. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.Y.X.; Van Langevelde, F.; Estrada-Peña, A.; Suzán, G.; de Boer, W.F. The diversity–disease relationship: Evidence for and criticisms of the dilution effect. Parasitology 2016, 143, 1075–1086. [Google Scholar] [CrossRef]
- Rubio, A.V.; Castro-Arellano, I.; Mills, J.N.; List, R.; Ávila-Flores, R.; Suzán, G. Is species richness driving intra- and interspecific interactions and temporal activity overlap of a hantavirus host? An experimental test. PLoS ONE 2017, 12, 1–19. [Google Scholar] [CrossRef]
- Wood, C.L.; McInturff, A.; Young, H.S.; Kim, D.; Lafferty, K.D. Human infectious disease burdens decrease with urbanization but not with biodiversity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160122. [Google Scholar] [CrossRef]
- Kilpatrick, A.M.; Salkeld, D.J.; Titcomb, G.; Hahn, M.B. Conservation of biodiversity as a strategy for improving human health and well-being. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160131. [Google Scholar] [CrossRef] [Green Version]
- LoGiudice, K.; Ostfeld, R.S.; Schmidt, K.A.; Keesing, F. The ecology of infectious disease: Effects of host diversity and community composition on Lyme disease risk. Proc. Natl. Acad. Sci. USA 2003, 100, 567–571. [Google Scholar] [CrossRef] [Green Version]
- Dizney, L.J.; Ruedas, L.A. Increased host species diversity and decreased prevalence of Sin Nombre virus. Emerg. Infect. Dis. 2009, 15, 1012. [Google Scholar] [CrossRef]
- Suzán, G.; Marcé, E.; Giermakowski, J.T.; Mills, J.N.; Ceballos, G.; Ostfeld, R.S.; Armién, B.; Pascale, J.M.; Yates, T.L. Experimental evidence for reduced rodent diversity causing increased hantavirus prevalence. PLoS ONE 2009, 4, e5461. [Google Scholar] [CrossRef]
- Suzán, G.; García-Peña, G.E.; Castro-Arellano, I.; Rico, O.; Rubio, A.V.; Tolsá, M.J.; Roche, B.; Hosseini, P.R.; Rizzoli, A.; Murray, K.A.; et al. Metacommunity and phylogenetic structure determine wildlife and zoonotic infectious disease patterns in time and space. Ecol. Evol. 2015, 5, 865–873. [Google Scholar] [CrossRef]
- McGill, B.J.; Enquist, B.J.; Weiher, E.; Westoby, M. Rebuilding community ecology from functional traits. Trends Ecol. Evol. 2006, 21, 178–185. [Google Scholar] [CrossRef]
- Keesing, F.; Belden, L.K.; Daszak, P.; Dobson, A.; Harvell, C.D.; Holt, R.D.; Hudson, P.; Jolles, A.; Jones, K.E.; Mitchell, C.E.; et al. Impacts of biodiversity on the emergence and transmission of infectious diseases. Nature 2010, 468, 647–652. [Google Scholar] [CrossRef]
- Herbreteau, V.; Gonzalez, J.P.; Hugot, J.P. Implication of phylogenetic systematics of Rodent-Borne hantaviruses allows understanding of their distribution. Ann. N. Y. Acad. Sci. 2006, 1081, 39–56. [Google Scholar] [CrossRef]
- Han, B.A.; Kramer, A.M.; Drake, J.M. Global patterns of zoonotic disease in mammals. Trends Parasitol. 2016, 32, 565–577. [Google Scholar] [CrossRef]
- Gosselin, F. Improving approaches to the analysis of functional and taxonomic biotic homogenization: Beyond mean specialization. J. Ecol. 2012, 100, 1289–1295. [Google Scholar] [CrossRef]
- Luan, V.D.; Yoshimatsu, K.; Endo, R.; Taruishi, M.; Huong, V.T.; Dat, D.T.; Tien, P.C.; Shimizu, K.; Koma, T.; Yasuda, S.P.; et al. Studies on hantavirus infection in small mammals captured in southern and central highland area of Vietnam. J. Vet. Med. Sci. 2012, 74, 1155–1162. [Google Scholar] [CrossRef]
- Xu, Z.Y.; Tang, Y.W.; Kan, L.Y.; Tsai, T.F. Cats-source of protection or infection? A case-control study of hemorrhagic fever with renal syndrome. Am. J. Epidemiol. 1987, 126, 942–948. [Google Scholar]
- Chin, C.; Chiueh, T.S.; Yang, W.C.; Yang, T.H.; Shih, C.M.; Lin, H.T.; Lin, K.C.; Lien, J.C.; Tsai, T.F.; Ruo, S.L.; et al. Hantavirus infection in Taiwan: The experience of a geographically unique area. J. Med. Virol. 2000, 60, 237–247. [Google Scholar] [CrossRef]
- Colombo, V.C.; Brignone, J.; Sen, C.; Previtali, M.A.; Martin, M.L.; Levis, S.; Monje, L.; González-Ittig, R.; Beldomenico, P.M. Orthohantavirus genotype Lechiguanas in Oligoryzomys nigripes (Rodentia: Cricetidae): New evidence of host-switching. Acta Trop. 2019, 191, 133–138. [Google Scholar] [CrossRef]
- Langlois, J.P.; Fahrig, L.; Merriam, G.; Artsob, H. Landscape structure influences continental distribution of hantavirus in deer mice. Landsc. Ecol. 2001, 16, 255–266. [Google Scholar] [CrossRef]
- Mackelprang, R.; Dearing, M.D.; St Jeor, S. High prevalence of Sin Nombre virus in rodent populations, central Utah: A consequence of human disturbance? Emerg. Infect. Dis. 2001, 7, 480–482. [Google Scholar] [CrossRef]
- Kuenzi, A.J.; Douglass, R.J.; White, D.J.; Bond, C.W.; Mills, J.N. Antibody to Sin Nombre virus in rodents associated with peridomestic habitats in west central Montana. Am. J. Trop. Med. Hyg. 2001, 64, 137–146. [Google Scholar] [CrossRef]
- Lonner, B.N.; Douglass, R.J.; Kuenzi, A.J.; Hughes, K. Seroprevalence against Sin Nombre virus in resident and dispersing deer mice. Vector-Borne Zoonotic Dis. 2008, 8, 433–442. [Google Scholar] [CrossRef]
- Calisher, C.H.; Mills, J.N.; Root, J.J.; Doty, J.B.; Beaty, B.J. The relative abundance of deer mice with antibody to Sin Nombre virus corresponds to the occurrence of Hantavirus Pulmonary Syndrome in nearby humans. Vector-Borne Zoonotic Dis. 2010, 11, 577–582. [Google Scholar] [CrossRef]
- McKinney, M.L. Urbanization as a major cause of biotic homogenization. Biol. Conserv. 2006, 127, 247–260. [Google Scholar] [CrossRef]
- Olden, J.D. Biotic homogenization: A new research agenda for conservation biogeography. J. Biogeogr. 2006, 33, 2027–2039. [Google Scholar] [CrossRef]
- Linske, M.A.; Williams, S.C.; Stafford, K.C.; Ortega, I.M. Ixodes scapularis (Acari: Ixodidae) reservoir host diversity and abundance impacts on dilution of Borrelia burgdorferi (Spirochaetales: Spirochaetaceae) in residential and woodland habitats in Connecticut, United States. J. Med. Entomol. 2018, 55, 681–690. [Google Scholar] [CrossRef]
- Yates, T.L.; Mills, J.N.; Parmenter, C.A.; Ksiazek, T.G.; Parmenter, R.R.; Vande Castle, J.R.; Calisher, C.H.; Nichol, S.T.; Abbott, K.D.; Young, J.C.; et al. The ecology and evolutionary history of an emergent disease: Hantavirus Pulmonary Syndrome. Bioscience 2006, 52, 989–998. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Model | df | logLik | AICc | Δ | Weight | R2 | p |
|---|---|---|---|---|---|---|---|
| tas~ (s ***) + (bp *) + (rank *) + (mntd **) | 6 | −577.401 | 1167.3 | 0.00 | 0.372 | 0.12 | <0.001 |
| tas~ (s **) + pie + (bp *) + (rank *) + (mntd **) | 7 | −577.152 | 1169.0 | 1.69 | 0.160 | 0.12 | <0.001 |
| tas~ (diversity2 ***) + (bp *) + rank + (mntd **) | 6 | −578.418 | 1169.4 | 2.03 | 0.135 | 0.11 | <0.001 |
| tas~ diversity2 + (bp *) + rank + (mntd *) + pd | 7 | −577.534 | 1168.8 | 2.45 | 0.109 | 0.11 | <0.001 |
| tas~ diversity2 + (bp *) + rank + (mntd *) + mpd + pd | 8 | −576.943 | 1170.8 | 3.48 | 0.065 | 0.11 | <0.001 |
| tas~ n + (s **) + pie + (bp *) + rank + (mntd **) | 8 | −576.955 | 1170.9 | 3.51 | 0.064 | 0.11 | <0.001 |
| tas~ n + (phylo1 **) + mntd | 5 | −580.731 | 1171.8 | 4.50 | 0.039 | 0.09 | <0.001 |
| tas~ n+(s **) + pie + (bp *) + rank + mpd + (mntd *) | 9 | −576.796 | 1172.8 | 5.43 | 0.025 | 0.11 | <0.001 |
| tas~ n + diversity1 + phylo1 + mntd | 6 | −580.493 | 1173.5 | 6.18 | 0.017 | 0.08 | 0.001 |
| tas~ n + s + pie + (bp *) + rank + mpd + pd + (mntd *) | 10 | −576.778 | 1175.0 | 7.67 | 0.008 | 0.1 | 0.002 |
| tas~ n + diversity1 + rank + phylo1 + mntd | 7 | −580.452 | 1175.6 | 8.29 | 0.006 | 0.08 | 0.003 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milholland, M.T.; Castro-Arellano, I.; Garcia-Peña, G.E.; Mills, J.N. The Ecology and Phylogeny of Hosts Drive the Enzootic Infection Cycles of Hantaviruses. Viruses 2019, 11, 671. https://doi.org/10.3390/v11070671
Milholland MT, Castro-Arellano I, Garcia-Peña GE, Mills JN. The Ecology and Phylogeny of Hosts Drive the Enzootic Infection Cycles of Hantaviruses. Viruses. 2019; 11(7):671. https://doi.org/10.3390/v11070671
Chicago/Turabian StyleMilholland, Matthew T., Iván Castro-Arellano, Gabriel E. Garcia-Peña, and James N. Mills. 2019. "The Ecology and Phylogeny of Hosts Drive the Enzootic Infection Cycles of Hantaviruses" Viruses 11, no. 7: 671. https://doi.org/10.3390/v11070671
APA StyleMilholland, M. T., Castro-Arellano, I., Garcia-Peña, G. E., & Mills, J. N. (2019). The Ecology and Phylogeny of Hosts Drive the Enzootic Infection Cycles of Hantaviruses. Viruses, 11(7), 671. https://doi.org/10.3390/v11070671