Novel Tick Phlebovirus Genotypes Lacking Evidence for Vertebrate Infections in Anatolia and Thrace, Turkey

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Approvals
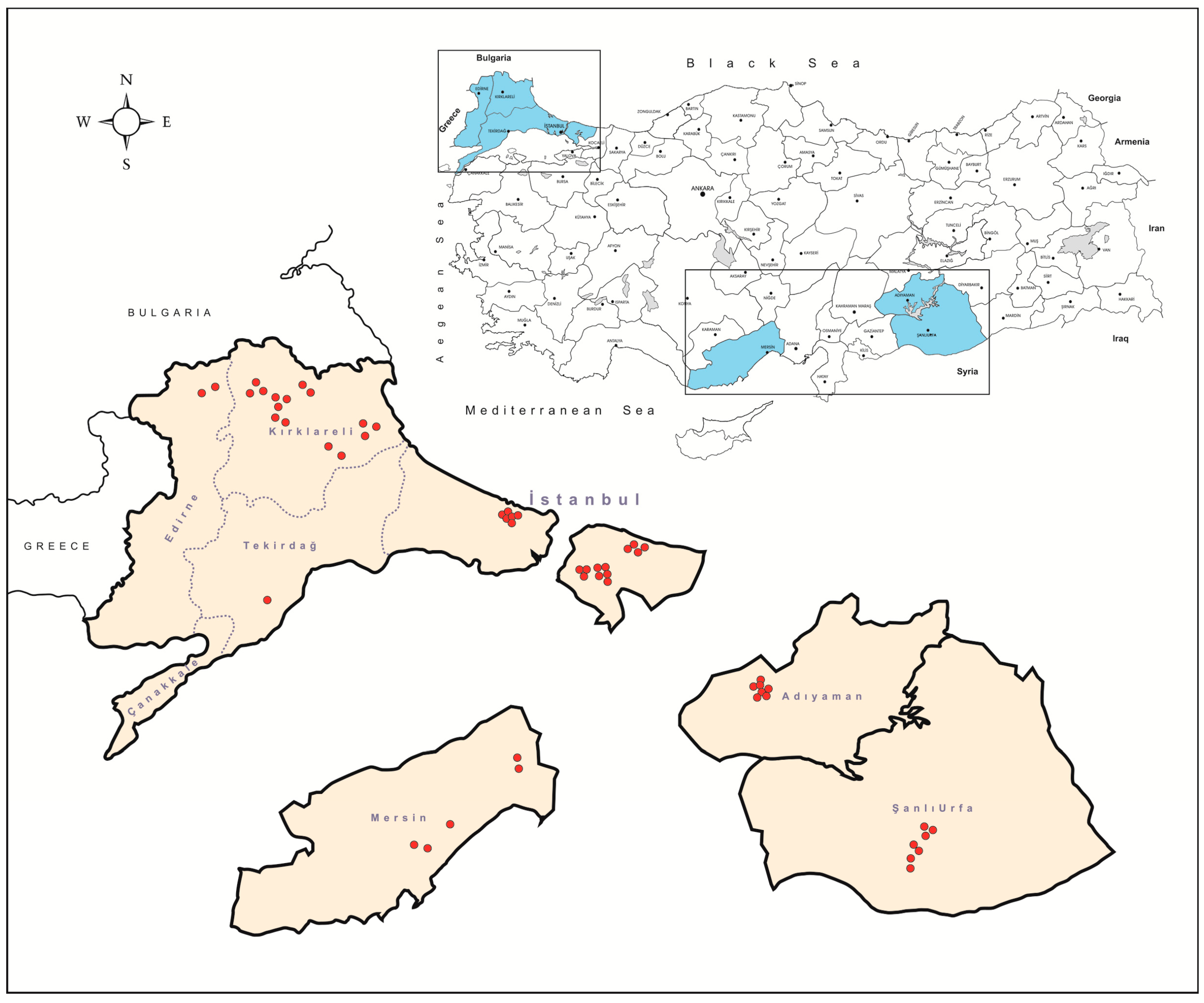
2.2. Tick Specimens
2.3. Specimen Processing
2.4. Virus Screening
2.5. Human Specimens
2.6. Virus Isolation
2.7. Sanger and Next Generation Sequencing (NGS)
2.8. Sequence Data Analysis
3. Results
3.1. The Tick Cohort and Screening Findings
3.2. Human Findings
3.3. Virus Isolation
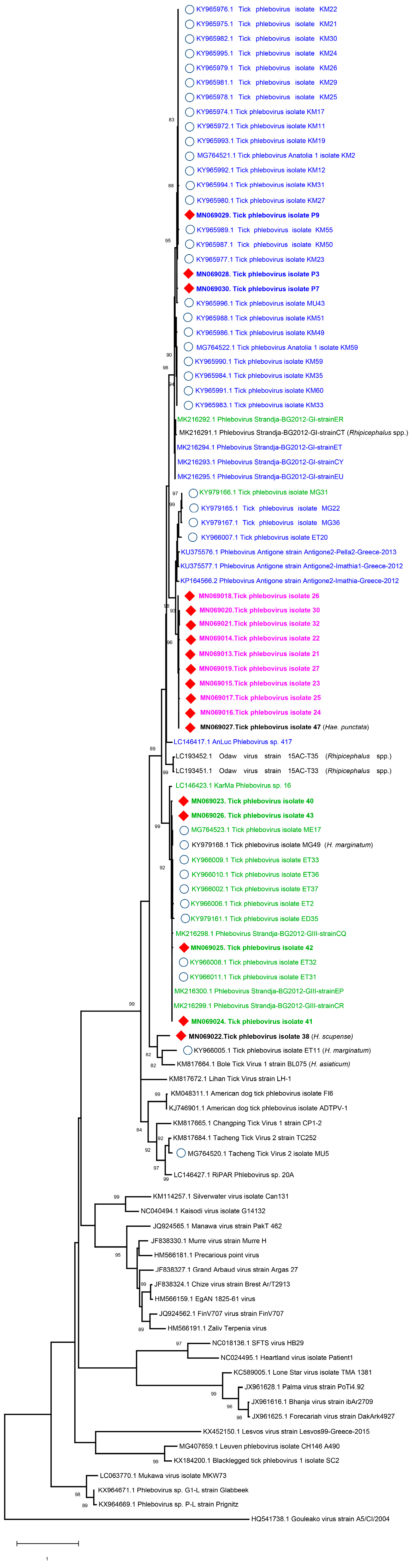
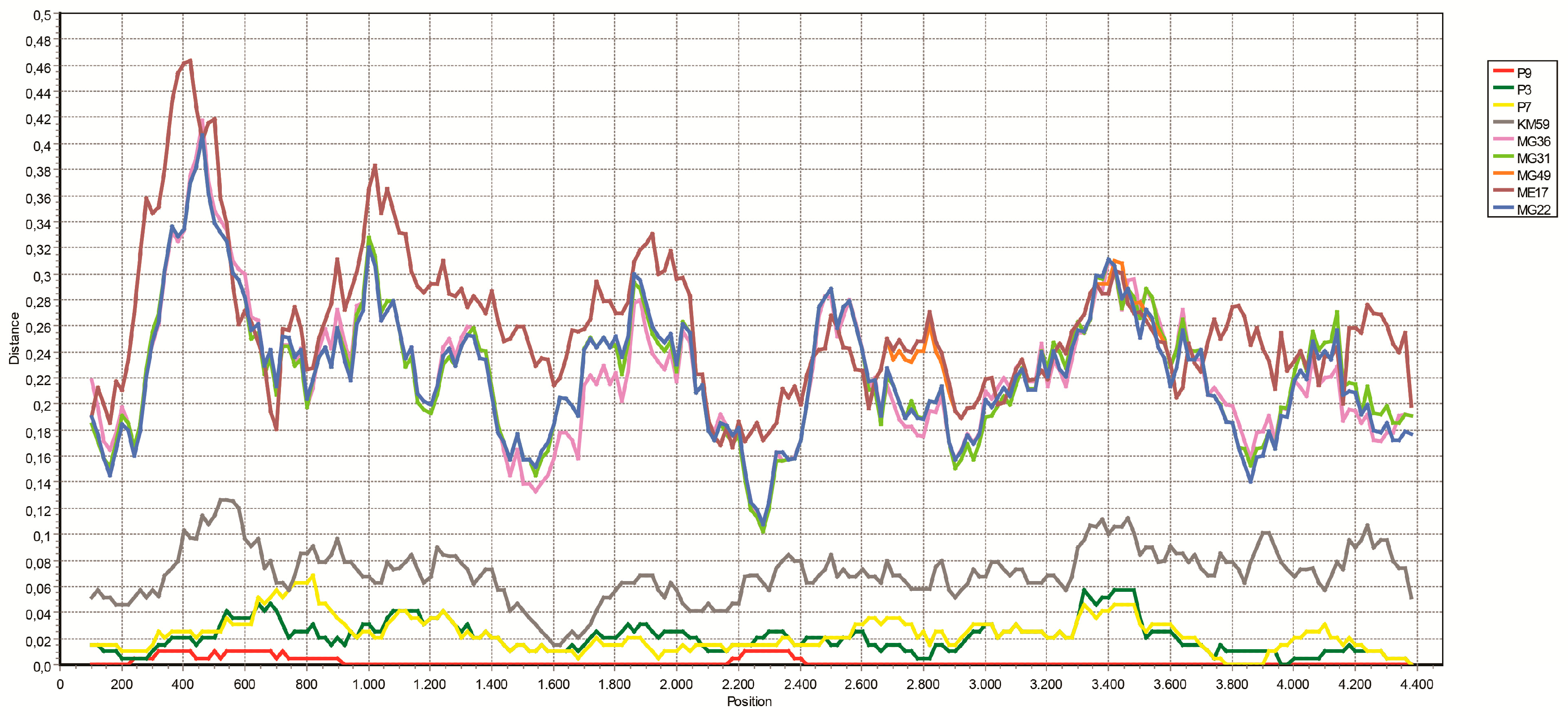
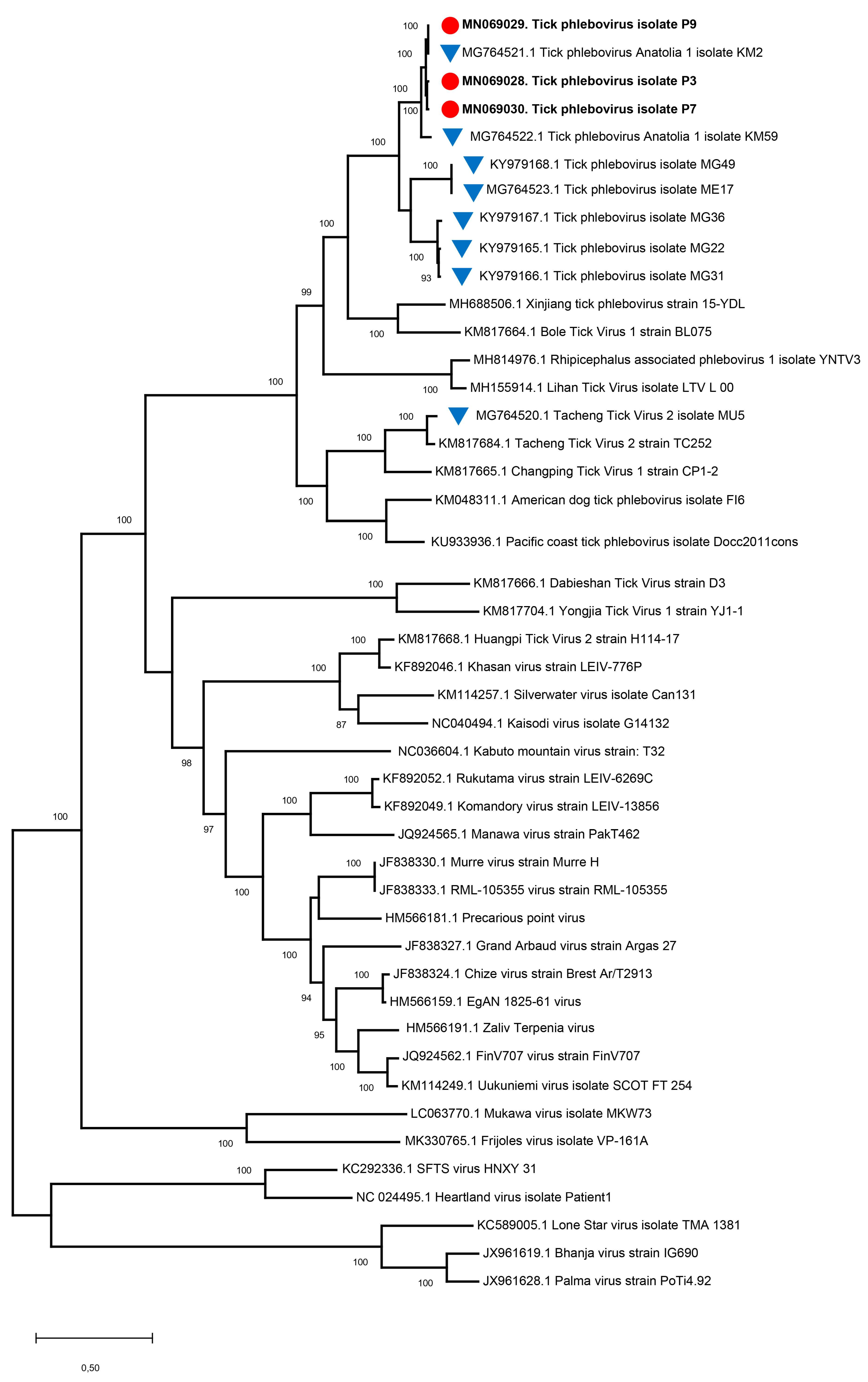
3.4. NGS Findings and Genomic Characterization of L Segments
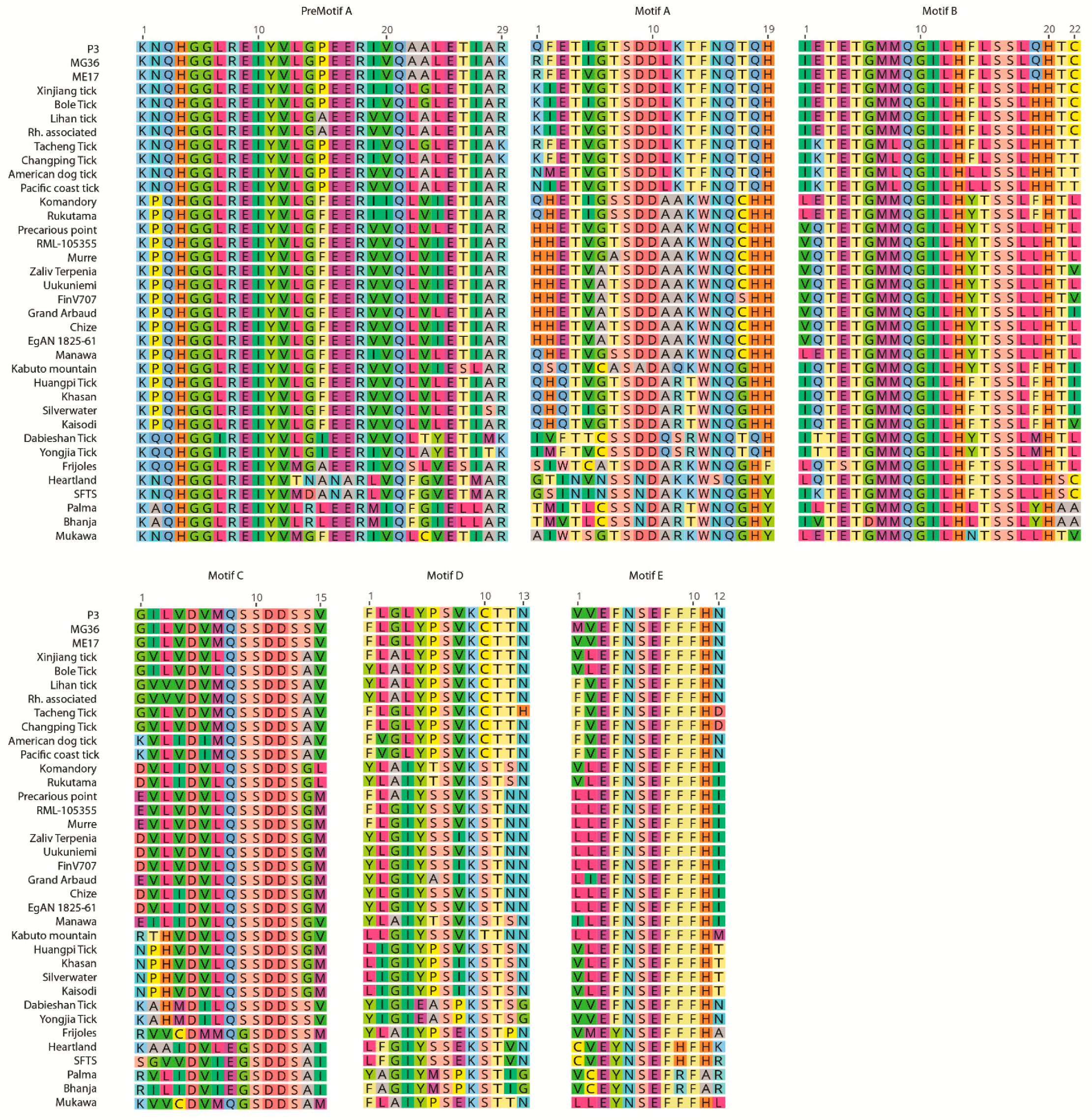
3.5. Analysis of Putative Coding Regions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Whitehouse, C.A.; Kuhn, J.H.; Wada, J.; Ergunay, K. Family Bunyaviridae. In Global Virology I: Identifying and Investigating Viral Diseases, 1st ed.; Shapshak, P., Sinnott, J.T., Somboonwit, C., Kuhn, J.H., Eds.; Springer: New York, NY, USA, 2015; pp. 199–246. [Google Scholar]
- Alkan, C.; Bichaud, L.; de Lamballerie, X.; Alten, B.; Gould, E.A.; Charrel, R.N. Sandfly-borne phleboviruses of Eurasia and Africa: Epidemiology, genetic diversity, geographic range, control measures. Antivir. Res. 2013, 100, 54–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Accardi, L.; Nicoletti, L.; Gro, M.C.; Takehara, K.; Hilditch, C.; Morikawa, S.; Bishop, D.H.L. Sequences and coding strategies of the S RNAs of Toscana and Rift Valley fever viruses compared to those of Punta Toro, Sicilian sandfly fever, and Uukuniemi viruses. Virology 1991, 180, 738–753. [Google Scholar] [CrossRef]
- Elliott, R.M.; Brennan, B. Emerging phleboviruses. Curr. Opin. Virol. 2014, 5, 50–57. [Google Scholar] [CrossRef] [Green Version]
- Abudurexiti, A.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Avšič-Županc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, É.; Blair, C.D.; et al. Taxonomy of the order Bunyavirales: Update 2019. Arch. Virol. 2019, 164, 1949–1965. [Google Scholar] [CrossRef] [PubMed]
- Silvas, J.A.; Aguilar, P.V. The emergence of severe fever with thrombocytopenia syndrome virus. Am. J. Trop. Med. Hyg. 2017, 97, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Brault, A.C.; Savage, H.M.; Duggal, N.K.; Eisen, R.J.; Staples, J.E. Heartland virus epidemiology, vector association, and disease potential. Viruses 2018, 10, E498. [Google Scholar] [CrossRef] [PubMed]
- Saikku, P. Arboviruses in Finland. 3. Uukuniemi virus antibodies in human, cattle, and reindeer sera. Am. J. Trop. Med. Hyg. 1973, 22, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Hubalek, Z. Biogeography of tick-borne bhanja virus (bunyaviridae) in Europe. Interdiscip. Perspect. Infect. Dis. 2009, 2009, 372691. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Weisend, C.; Kajihara, M.; Matysiak, C.; Williamson, B.N.; Simuunza, M.; Mweene, A.S.; Takada, A.; Tesh, R.B.; Ebihara, H. Comprehensive molecular detection of tick-borne phleboviruses leads to the retrospective identification of taxonomically unassigned bunyaviruses and the discovery of a novel member of the genus phlebovirus. J. Virol. 2015, 89, 594–604. [Google Scholar] [CrossRef]
- Brinkmann, A.; Dinçer, E.; Polat, C.; Hekimoğlu, O.; Hacıoğlu, S.; Földes, K.; Özkul, A.; Öktem, İ.M.A.; Nitsche, A.; Ergünay, K. A metagenomic survey identifies Tamdy orthonairovirus as well as divergent phlebo-, rhabdo-, chu- and flavi-like viruses in Anatolia, Turkey. Ticks Tick Borne Dis. 2018, 9, 1173–1183. [Google Scholar] [CrossRef]
- Matsuno, K.; Kajihara, M.; Nakao, R.; Nao, N.; Mori-Kajihara, A.; Muramatsu, M.; Qiu, Y.; Torii, S.; Igarashi, M.; Kasajima, N.; et al. The unique phylogenetic position of a novel tick-borne phlebovirus ensures an Ixodid origin of the genus Phlebovirus. mSphere 2018, 3, e00239-18. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLlife 2015, 4, e05378. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.; Figueira, L.; Nunes, M.; Esteves, A.; Cotao, A.J.; Vieira, M.L.; Maia, C.; Campino, L.; Parreira, R. Multiple Phlebovirus (Bunyaviridae) genetic groups detected in Rhipicephalus, Hyalomma and Dermacentor ticks from southern Portugal. Ticks Tick Borne Dis. 2017, 8, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Kontana, A.; Tsioka, K.; Chaligiannis, I.; Sotiraki, S. Novel phleboviruses detected in ticks, Greece. Ticks Tick Borne Dis. 2016, 7, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Kontana, A.; Tsioka, K.; Saratsis, A.; Sotiraki, S. Novel phlebovirus detected in Haemaphysalis parva ticks in a Greek island. Ticks Tick Borne Dis. 2017, 8, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Ohlendorf, V.; Marklewitz, M.; Kopp, A.; Yordanov, S.; Drosten, C.; Junglen, S. Huge diversity of phleboviruses in ticks from Strandja Nature Park, Bulgaria. Ticks Tick Borne Dis. 2019, 10, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Dinçer, E.; Brinkmann, A.; Hekimoğlu, O.; Hacıoğlu, S.; Földes, K.; Karapınar, Z.; Polat, P.F.; Oğuz, B.; Orunç Kılınç, Ö.; Hagedorn, P.; et al. Generic amplification and next generation sequencing reveal Crimean-Congo hemorrhagic fever virus AP92-like strain and distinct tick phleboviruses in Anatolia, Turkey. Parasites Vectors 2017, 10, 335. [Google Scholar] [CrossRef] [PubMed]
- Filippova, N.A. Fauna of Russia and Neighbouring Countries. Ixodid Ticks of Subfamily Amblyomminae; Nauka Publishing House: St. Petersburg, Russia, 1997. [Google Scholar]
- Walker, J.B.; Keirans, J.E.; Horak, I.G. The Genus Rhipicephalus (Acari, Ixodidae): A Guide to the Brown Ticks of the World, Rev. ed.; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Walker, A.R.; Bouattour, A.; Camicas, J.L.; Estrada-Pena, A.; Horak, I.G.; Latif, A.A.; Pegram, R.G.; Preston, P.M. Ticks of Domestic Animals in Africa: A Guide to Identification of Species, 1st ed.; Bioscience Reports: Edinburgh, UK, 2003. [Google Scholar]
- Estrada-Peña, A.; Bouattour, A.; Camicas, J.L.; Walker, A.R. Ticks of Domestic Animals in the Mediterranean Region, 1st ed.; University of Zaragoza Press: Zaragoza, Spain, 2004. [Google Scholar]
- Apanaskevich, D.A.; Horak, I.G. The genus Hyalomma Koch, 1844: V. Reevaluation of the taxonomic rank of taxa comprising the H. (Euhyalomma) marginatum Koch complex of species (Acari: Ixodidae) with redescription of all parasitic stages and notes on biology. Int. J. Acarol. 2008, 34, 13–42. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Herbig, A.; Maixner, F.; Bos, K.I.; Zink, A.; Krause, J.; Huson, D.H. MALT: Fast alignment and analysis of metagenomic DNA sequence data applied to the Tyrolean Iceman. bioRxiv 2016. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Gorska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN community edition—Interactive exploration and analysis of large-scale microbiome sequencing data. PLoS. Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Bateman, A.; Birney, E.; Cerruti, L.; Durbin, R.; Etwiller, L.; Eddy, S.R.; Griffiths-Jones, S.; Howe, K.L.; Marshall, M.; Sonnhammer, E.L. The Pfam protein families database. Nucleic Acids Res. 2002, 30, 276–280. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, 222–226. [Google Scholar] [CrossRef]
- Palacios, G.; Savji, N.; Travassos da Rosa, A.; Guzman, H.; Yu, X.; Desai, A.; Rosen, G.E.; Hutchison, S.; Lipkin, W.I.; Tesh, R. Characterization of the Uukuniemi virus group (Phlebovirus: Bunyaviridae): Evidence for seven distinct species. J. Virol. 2013, 87, 3187–3195. [Google Scholar] [CrossRef]
- Muller, R.; Poch, O.; Delarue, M.; Bishop, D.H.; Bouloy, M. Rift Valley fever virus L segment: Correction of the sequence and possible functional role of newly identified regions conserved in RNA-dependent polymerases. J. Gen. Virol. 1994, 75, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Poch, O.; Sauvaget, I.; Delarue, M.; Tordo, N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 1989, 8, 3867–3874. [Google Scholar] [CrossRef] [PubMed]
- Aquino, V.H.; Moreli, M.L.; Moraes Figueiredo, L.T. Analysis of oropouche virus L protein amino acid sequence showed the presence of an additional conserved region that could harbour an important role for the polymerase activity. Arch. Virol. 2003, 148, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, V.; Afonso, R.; Nunes, M.; Vieira, M.L.; Bravo-Barriga, D.; Frontera, E.; Martinez, M.; Pereira, A.; Maia, C.; Paiva-Cardoso, M.D.N.; et al. Geographic dispersal and genetic diversity of tick-borne phleboviruses (Phenuiviridae, Phlebovirus) as revealed by the analysis of L segment sequences. Ticks Tick Borne Dis. 2019, 10, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Fuchs, J.; Ehrmann, S.; Scherer-Lorenzen, M.; Kochs, G.; Panning, M. Molecular identification of novel phlebovirus sequences in European ticks. Ticks Tick Borne Dis. 2017, 8, 795–798. [Google Scholar] [CrossRef]
- Bouquet, J.; Melgar, M.; Swei, A.; Delwart, E.; Lane, R.S.; Chiu, C.Y. Metagenomic-based surveillance of Pacific coast tick Dermacentor occidentalis identifies two novel Bunyaviruses and an emerging human Ricksettsial pathogen. Sci. Rep. 2017, 7, 12234. [Google Scholar] [CrossRef]
- Tokarz, R.; Williams, S.H.; Sameroff, S.; Sanchez Leon, M.; Jain, K.; Lipkin, W.I. Virome analysis of Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis ticks reveals novel highly divergent vertebrate and invertebrate viruses. J. Virol. 2014, 88, 11480–11492. [Google Scholar] [CrossRef]
- Souza, W.M.; Fumagalli, M.J.; Torres Carrasco, A.O.; Romeiro, M.F.; Modha, S.; Seki, M.C.; Gheller, J.M.; Daffre, S.; Nunes, M.R.T.; Murcia, P.R.; et al. Viral diversity of Rhipicephalus microplus parasitizing cattle in southern Brazil. Sci. Rep. 2018, 8, 16315. [Google Scholar] [CrossRef]
- Ejiri, H.; Lim, C.K.; Isawa, H.; Yamaguchi, Y.; Fujita, R.; Takayama-Ito, M.; Kuwata, R.; Kobayashi, D.; Horiya, M.; Posadas-Herrera, G.; et al. Isolation and characterization of Kabuto Mountain virus, a new tick-borne phlebovirus from Haemaphysalis flava ticks in Japan. Virus Res. 2018, 244, 252–261. [Google Scholar] [CrossRef]
- Shen, S.; Duan, X.; Wang, B.; Zhu, L.; Zhang, Y.; Zhang, J.; Wang, J.; Luo, T.; Kou, C.; Liu, D.; et al. A novel tick-borne phlebovirus, closely related to severe fever with thrombocytopenia syndrome virus and Heartland virus, is a potential pathogen. Emerg. Microbes Infect. 2018, 7, 95. [Google Scholar] [CrossRef]
- Liu, M.M.; Lei, X.Y.; Yu, X.J. Meta-analysis of the clinical and laboratory parameters of SFTS patients in China. Virol. J. 2016, 13, 198. [Google Scholar] [CrossRef] [Green Version]
- Cui, N.; Liu, R.; Lu, Q.B.; Wang, L.Y.; Qin, S.L.; Yang, Z.D.; Zhuang, L.; Liu, K.; Li, H.; Zhang, X.A.; et al. Severe fever with thrombocytopenia syndrome bunyavirus-related human encephalitis. J. Infect. 2015, 70, 52–59. [Google Scholar] [CrossRef]
- Kim, U.J.; Kim, D.M.; Ahn, J.H.; Kang, S.J.; Jang, H.C.; Park, K.H.; Jung, S.I. Successful treatment of rapidly progressing severe fever with thrombocytopenia syndrome with neurological complications using intravenous immunoglobulin and corticosteroid. Antivir. Ther. 2016, 21, 637–640. [Google Scholar] [CrossRef] [Green Version]
- Calisher, C.H.; Goodpasture, H.C. Human infection with Bhanja virus. Am. J. Trop. Med. Hyg. 1975, 24, 1040–1042. [Google Scholar] [CrossRef]
- Vesenjak-Hirjan, J.; Calisher, C.H.; Beus, I.; Marton, E. First natural clinical human Bhanja virus infection. In Arboviruses in the Mediterranean Countries: 6th FEMS Symposium, 1st ed.; Vesenjak-Hirjan, J., Porterfield, J.S., Arslanagıc, E., Eds.; Fischer: Stuttgart, Germany, 1980; pp. 297–301. [Google Scholar]
- Rezelj, V.V.; Mottram, T.J.; Hughes, J.; Elliott, R.M.; Kohl, A.; Brennan, B. M segment-based minigenomes and virus-like particle assays as an approach to assess the potential of tickborne Phlebovirus genome reassortment. J. Virol. 2019, 93, e02068-18. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Province | Species | Pools (n) | Positive Pool (n/%) | Infection Rate (%) |
|---|---|---|---|---|
| Mersin | R. bursa (n = 65) | 11 | 2/18.2 | 3.1 |
| R. sanguineus s.l. (n = 117) | 14 | 3/21.4 | 2.6 | |
| Şanlıurfa | R. sanguineus s.l. (n = 67) | 10 | 2/20 | 2.9 |
| Tekirdağ | R. turanicus (n = 264) | 12 | 5/41.6 | 1.9 |
| Kırklareli | R. bursa (n = 438) | 55 | 4/7.2 | 0.9 |
| R. turanicus (n = 319) | 42 | 6/14.3 | 1.9 | |
| Hae. punctata (n = 73) | 15 | 2/13.3 | 2.7 | |
| H. scupense (n = 962) | 49 | 2/4.1 | 0.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emanet, N.; Kar, S.; Dinçer, E.; Brinkmann, A.; Hacıoğlu, S.; Farzani, T.A.; Koçak Tufan, Z.; Polat, P.F.; Şahan, A.; Özkul, A.; et al. Novel Tick Phlebovirus Genotypes Lacking Evidence for Vertebrate Infections in Anatolia and Thrace, Turkey. Viruses 2019, 11, 703. https://doi.org/10.3390/v11080703
Emanet N, Kar S, Dinçer E, Brinkmann A, Hacıoğlu S, Farzani TA, Koçak Tufan Z, Polat PF, Şahan A, Özkul A, et al. Novel Tick Phlebovirus Genotypes Lacking Evidence for Vertebrate Infections in Anatolia and Thrace, Turkey. Viruses. 2019; 11(8):703. https://doi.org/10.3390/v11080703
Chicago/Turabian StyleEmanet, Nergis, Sırrı Kar, Ender Dinçer, Annika Brinkmann, Sabri Hacıoğlu, Touraj Aligholipour Farzani, Zeliha Koçak Tufan, Pelin Fatoş Polat, Adem Şahan, Aykut Özkul, and et al. 2019. "Novel Tick Phlebovirus Genotypes Lacking Evidence for Vertebrate Infections in Anatolia and Thrace, Turkey" Viruses 11, no. 8: 703. https://doi.org/10.3390/v11080703
APA StyleEmanet, N., Kar, S., Dinçer, E., Brinkmann, A., Hacıoğlu, S., Farzani, T. A., Koçak Tufan, Z., Polat, P. F., Şahan, A., Özkul, A., Nitsche, A., Linton, Y. -M., & Ergünay, K. (2019). Novel Tick Phlebovirus Genotypes Lacking Evidence for Vertebrate Infections in Anatolia and Thrace, Turkey. Viruses, 11(8), 703. https://doi.org/10.3390/v11080703