Genome of Alaskapox Virus, a Novel Orthopoxvirus Isolated from Alaska

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Sequences Used for Analysis
2.2. NGS Sequencing, De Novo Assembly and Gap Filling
2.3. Gene Prediction and Annotation
2.4. Alignment of Poxvirus Genomes
2.5. Phylogenetic Analysis of the Conserved Central region
2.6. Gene Content Comparison
2.7. dN/dS Analysis
2.8. Investigation of Potential Recombination Events
3. Results
3.1. Genome Characteristics
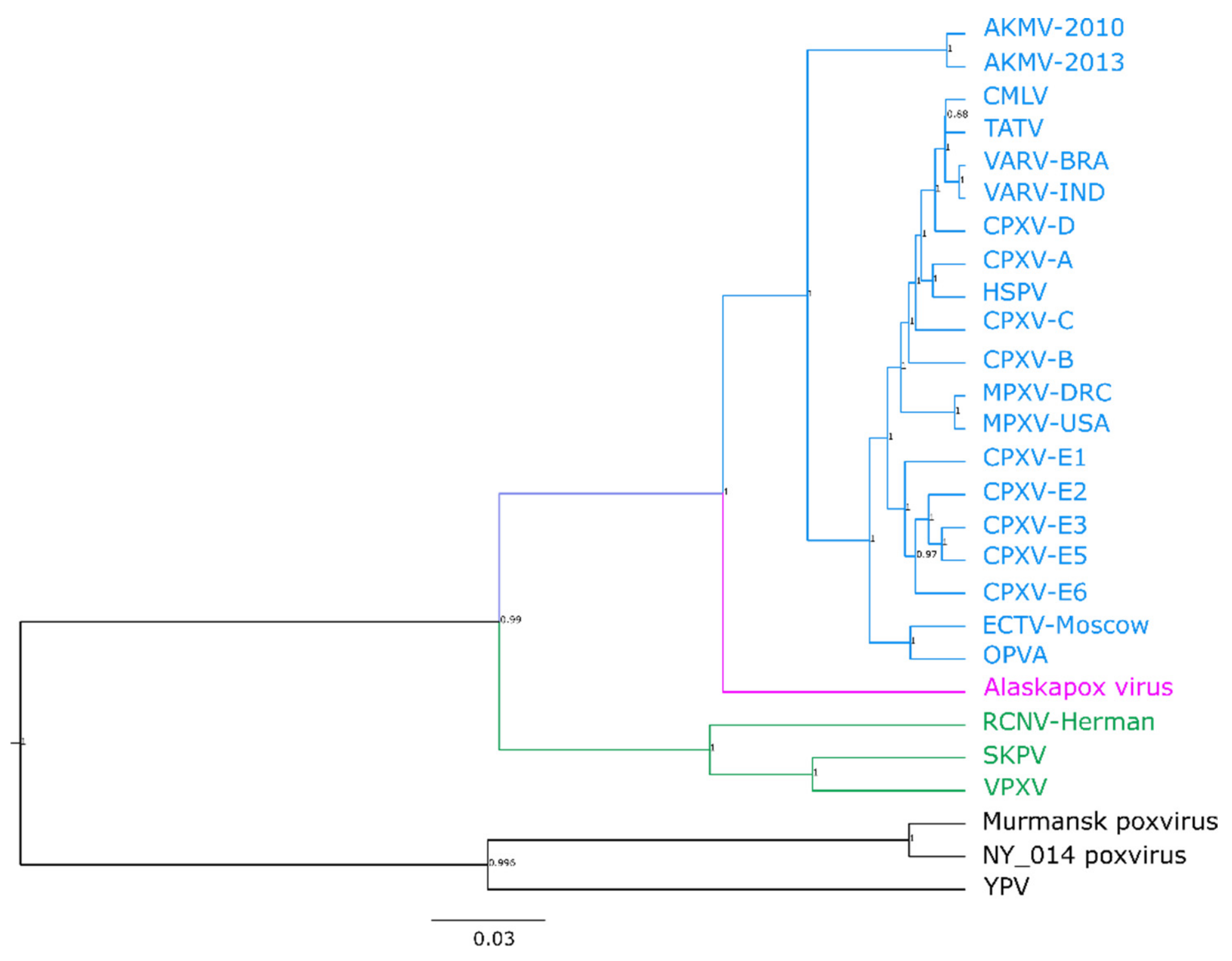
3.2. Phylogenetic Analysis of the Central Core Region
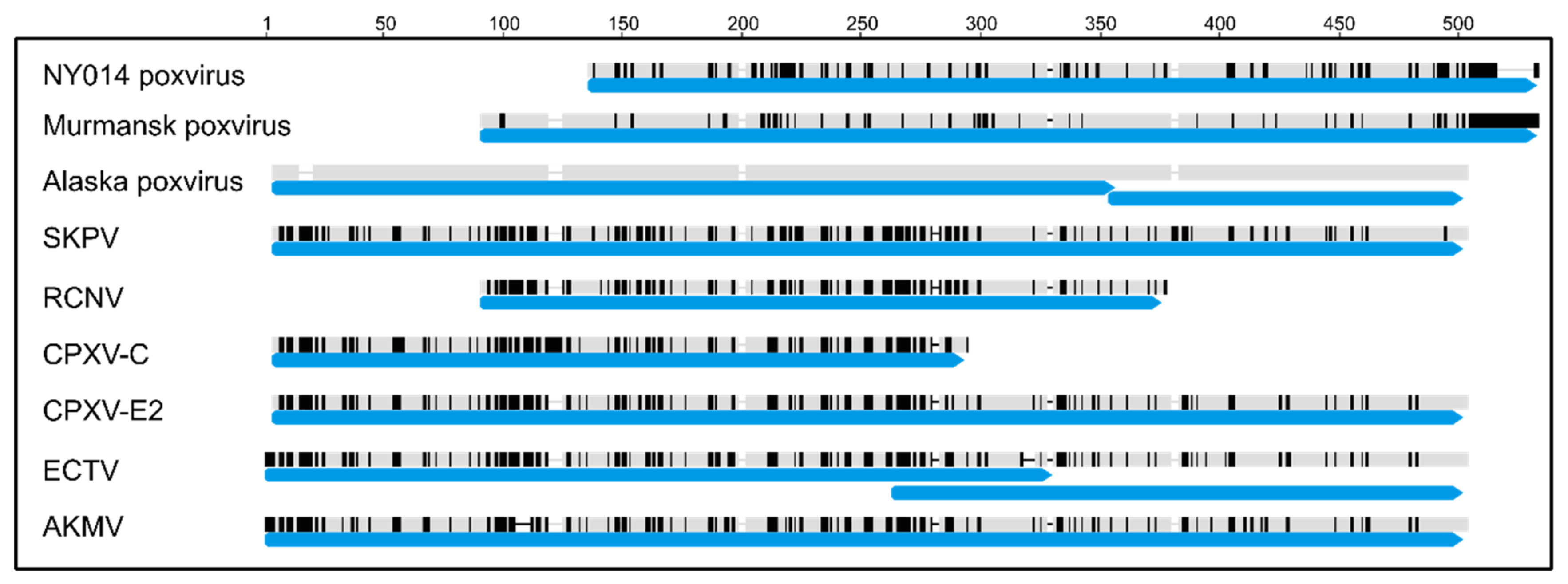
3.3. Comparison of Gene Content
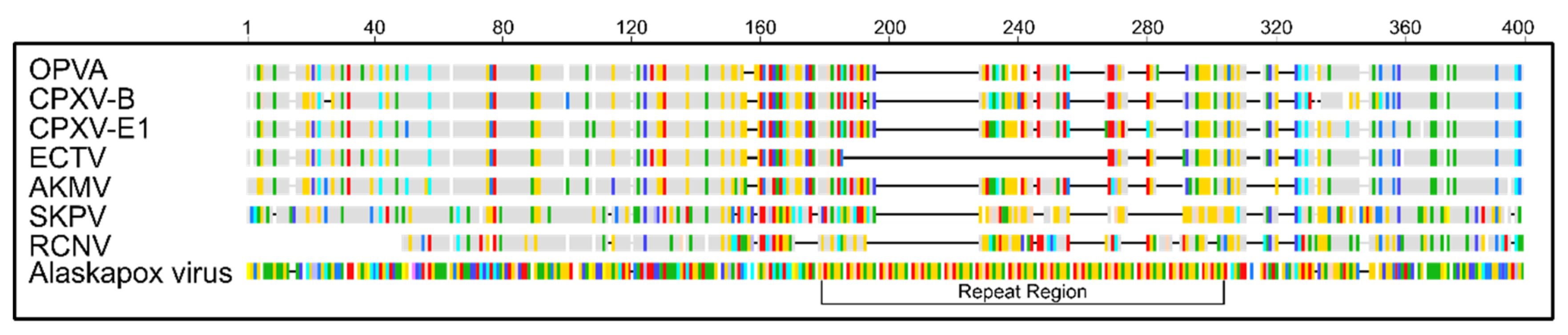
3.4. Comparison of Putative Host Range/Virulence Factors
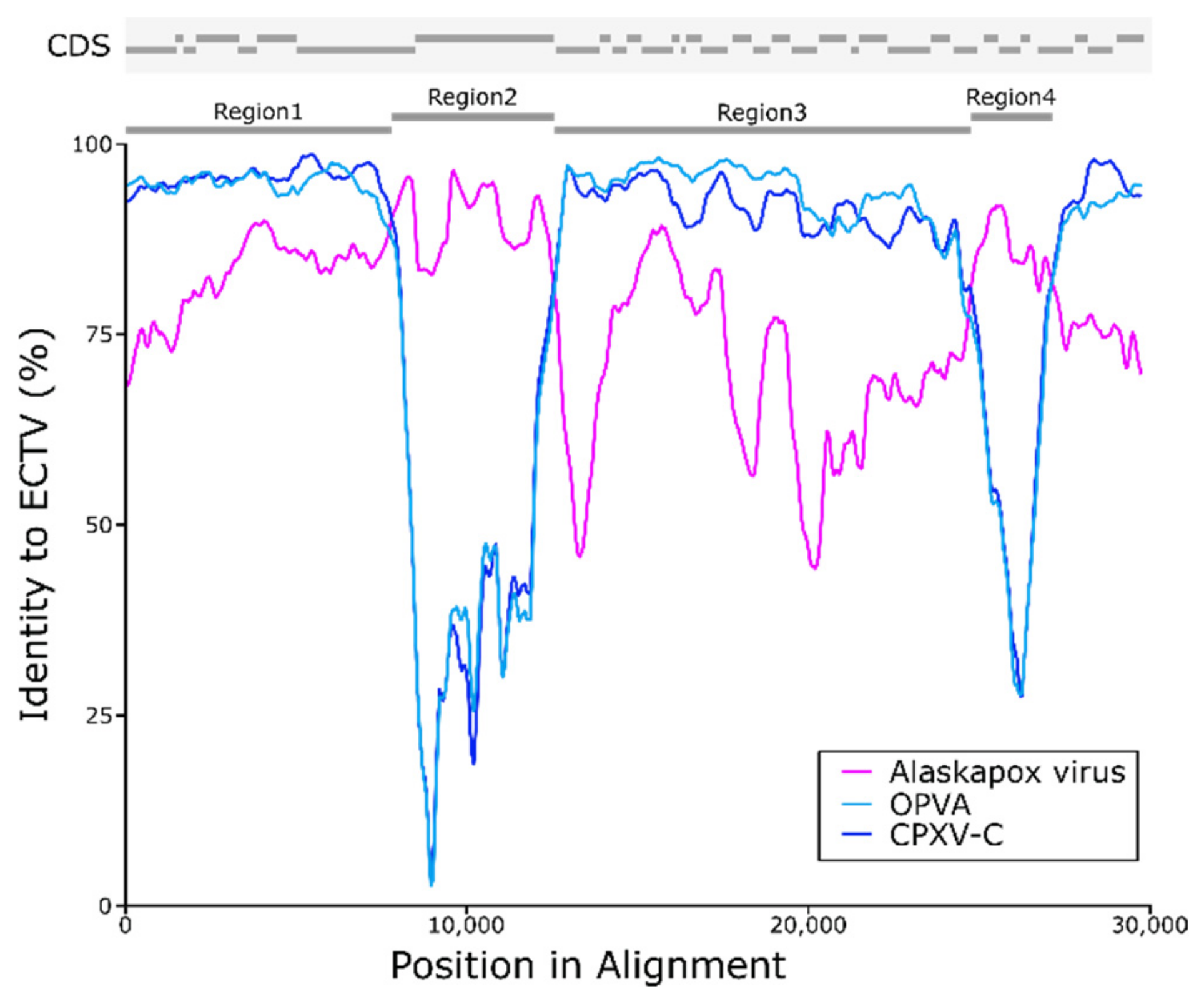
3.5. Possible Recombination with Ectromelia Virus (ECTV)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Disclosure
Conflicts of Interest
References
- Haller, S.L.; Peng, C.; McFadden, G.; Rothenburg, S. Poxviruses and the evolution of host range and virulence. Infect. Genet. Evol. 2014, 21, 15–40. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.G.; Guagliardo, S.A.J.; Nakazawa, Y.J.; Doty, J.B.; Mauldin, M.R. Understanding orthopoxvirus host range and evolution: From the enigmatic to the usual suspects. Curr. Opin. Virol. 2018, 28, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Essbauer, S.; Pfeffer, M.; Meyer, H. Zoonotic poxviruses. Vet. Microbiol. 2010, 140, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Vorou, R.M.; Papavassiliou, V.G.; Pierroutsakos, I.N. Cowpox virus infection: An emerging health threat. Curr. Opin. Infect. Dis. 2008, 21, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, A.; Pauli, G. Sporadic human cases of cowpox in Germany. Eurosurveillance 2007, 12, pii=3178. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, D.B.; Eckburg, P.B. Human monkeypox: An emerging zoonosis. Lancet Infect. Dis. 2004, 4, 15–25. [Google Scholar] [CrossRef]
- Rimoin, A.W.; Mulembakani, P.M.; Johnston, S.C.; Lloyd Smith, J.O.; Kisalu, N.K.; Kinkela, T.L.; Blumberg, S.; Thomassen, H.A.; Pike, B.L.; Fair, J.N.; et al. Major increase in human monkeypox incidence 30 years after smallpox vaccination campaigns cease in the Democratic Republic of Congo. Proc. Natl. Acad. Sci. USA 2010, 107, 16262–16267. [Google Scholar] [CrossRef] [Green Version]
- Nolen, L.D.; Osadebe, L.; Katomba, J.; Likofata, J.; Mukadi, D.; Monroe, B.; Doty, J.; Kalemba, L.; Malekani, J.; Kabamba, J.; et al. Introduction of Monkeypox into a Community and Household: Risk Factors and Zoonotic Reservoirs in the Democratic Republic of the Congo. Am. J. Trop. Med. Hyg. 2015, 93, 410–415. [Google Scholar] [CrossRef] [Green Version]
- Kalthan, E.; Tenguere, J.; Ndjapou, S.G.; Koyazengbe, T.A.; Mbomba, J.; Marada, R.M.; Rombebe, P.; Yangueme, P.; Babamingui, M.; Sambella, A.; et al. Investigation of an outbreak of monkeypox in an area occupied by armed groups, Central African Republic. Med. Mal. Infect. 2018, 48, 263–268. [Google Scholar] [CrossRef]
- Kantele, A.; Chickering, K.; Vapalahti, O.; Rimoin, A.W. Emerging diseases-the monkeypox epidemic in the Democratic Republic of the Congo. Clin. Microbiol. Infect. 2016, 22, 658–659. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Multistate outbreak of monkeypox—Illinois, Indiana, and Wisconsin, 2003. Morb. Mortal. Wkly. Rep. 2003, 52, 537–540. [Google Scholar]
- Nagasse-Sugahara, T.K.; Kisielius, J.J.; Ueda-Ito, M.; Curti, S.P.; Figueiredo, C.A.; Cruz, A.S.; Silva, M.M.; Ramos, C.H.; Silva, M.C.; Sakurai, T.; et al. Human vaccinia-like virus outbreaks in Sao Paulo and Goias States, Brazil: Virus detection, isolation and identification. Rev. Inst. Med. Trop Sao Paulo 2004, 46, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Damaso, C.R.; Esposito, J.J.; Condit, R.C.; Moussatche, N. An emergent poxvirus from humans and cattle in Rio de Janeiro State: Cantagalo virus may derive from Brazilian smallpox vaccine. Virology 2000, 277, 439–449. [Google Scholar] [CrossRef]
- Abrahao, J.S.; Campos, R.K.; Trindade Gde, S.; Guimaraes da Fonseca, F.; Ferreira, P.C.; Kroon, E.G. Outbreak of severe zoonotic vaccinia virus infection, Southeastern Brazil. Emerg. Infect. Dis. 2015, 21, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Leite, J.A.; Drumond, B.P.; Trindade, G.S.; Lobato, Z.I.; da Fonseca, F.G.; Dos, S.J.; Madureira, M.C.; Guedes, M.I.; Ferreira, J.M.; Bonjardim, C.A.; et al. Passatempo virus, a vaccinia virus strain, Brazil. Emerg. Infect. Dis. 2005, 11, 1935–1938. [Google Scholar] [CrossRef]
- Erez, N.; Achdout, H.; Milrot, E.; Schwartz, Y.; Wiener-Well, Y.; Paran, N.; Politi, B.; Tamir, H.; Israely, T.; Weiss, S.; et al. Diagnosis of Imported Monkeypox, Israel, 2018. Emerg. Infect. Dis. 2019, 25, 980–983. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, A.; Aarons, E.; Astbury, J.; Balasegaram, S.; Beadsworth, M.; Beck, C.R.; Chand, M.; O’Connor, C.; Dunning, J.; Ghebrehewet, S.; et al. Two cases of monkeypox imported to the United Kingdom, September 2018. Eurosurveillance 2018, 23. [Google Scholar] [CrossRef]
- Singapore Ministry of Health, Confirmed Imported Case of Monkeypox in Singapore. Singapore Government: 9 May 2019. Available online: https://www.moh.gov.sg/news-highlights/details/confirmed-imported-case-of-monkeypox-in-singapore (accessed on 15 July 2019).
- Mombouli, J.V.; Ostroff, S.M. The remaining smallpox stocks: The healthiest outcome. Lancet 2012, 379, 10–12. [Google Scholar] [CrossRef]
- Fenner, F. The global eradication of smallpox. Med. J. Aust. 1980, 1, 916–930. [Google Scholar] [CrossRef]
- Reynolds, M.G.; Damon, I.K. Outbreaks of human monkeypox after cessation of smallpox vaccination. Trends Microbiol. 2012, 20, 80–87. [Google Scholar] [CrossRef]
- Vora, N.M.; Li, Y.; Geleishvili, M.; Emerson, G.L.; Khmaladze, E.; Maghlakelidze, G.; Navdarashvili, A.; Zakhashvili, K.; Kokhreidze, M.; Endeladze, M.; et al. Human infection with a zoonotic orthopoxvirus in the country of Georgia. N. Engl. J. Med. 2015, 372, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Lakis, N.S.; Li, Y.; Abraham, J.L.; Upton, C.; Blair, D.C.; Smith, S.; Zhao, H.; Damon, I.K. Novel Poxvirus Infection in an Immune Suppressed Patient. Clin. Infect. Dis. 2015, 61, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Smithson, C.; Meyer, H.; Gigante, C.M.; Gao, J.; Zhao, H.; Batra, D.; Damon, I.; Upton, C.; Li, Y. Two novel poxviruses with unusual genome rearrangements: NY_014 and Murmansk. Virus Genes 2017, 53, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Franke, A.; Jenckel, M.; Tamosiunaite, A.; Schluckebier, J.; Granzow, H.; Hoffmann, B.; Fischer, S.; Ulrich, R.G.; Hoper, D.; et al. Out of the Reservoir: Phenotypic and Genotypic Characterization of a Novel Cowpox Virus Isolated from a Common Vole. J. Virol. 2015, 89, 10959–10969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osadebe, L.U.; Manthiram, K.; McCollum, A.M.; Li, Y.; Emerson, G.L.; Gallardo-Romero, N.F.; Doty, J.B.; Wilkins, K.; Zhao, H.; Drew, C.P.; et al. Novel poxvirus infection in 2 patients from the United States. Clin. Infect. Dis. 2015, 60, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Gigante, C.; Khmaladze, E.; Liu, P.; Tang, S.; Wilkins, K.; Zhao, K.; Davidson, W.; Nakazawa, Y.; Maghlakelidze, G.; et al. Genome Sequences of Akhmeta Virus, an Early Divergent Old World Orthopoxvirus. Viruses 2018, 10, 252. [Google Scholar] [CrossRef]
- Cardeti, G.; Gruber, C.E.M.; Eleni, C.; Carletti, F.; Castilletti, C.; Manna, G.; Rosone, F.; Giombini, E.; Selleri, M.; Lapa, D.; et al. Fatal Outbreak in Tonkean Macaques Caused by Possibly Novel Orthopoxvirus, Italy, January 2015. Emerg. Infect. Dis. 2017, 23, 1941–1949. [Google Scholar] [CrossRef]
- Gruber, C.E.M.; Giombini, E.; Selleri, M.; Tausch, S.H.; Andrusch, A.; Tyshaieva, A.; Cardeti, G.; Lorenzetti, R.; De Marco, L.; Carletti, F.; et al. Whole Genome Characterization of Orthopoxvirus (OPV) Abatino, a Zoonotic Virus Representing a Putative Novel Clade of Old World Orthopoxviruses. Viruses 2018, 10, 546. [Google Scholar] [CrossRef]
- Springer, Y.P.; Hsu, C.H.; Werle, Z.R.; Olson, L.E.; Cooper, M.P.; Castrodale, L.J.; Fowler, N.; McCollum, A.M.; Goldsmith, C.S.; Emerson, G.L.; et al. Novel Orthopoxvirus Infection in an Alaska Resident. Clin. Infect. Dis. 2017, 64, 1737–1741. [Google Scholar] [CrossRef]
- Lanave, G.; Dowgier, G.; Decaro, N.; Albanese, F.; Brogi, E.; Parisi, A.; Losurdo, M.; Lavazza, A.; Martella, V.; Buonavoglia, C.; et al. Novel Orthopoxvirus and Lethal Disease in Cat, Italy. Emerg. Infect. Dis. 2018, 24, 1665–1673. [Google Scholar] [CrossRef] [Green Version]
- Skinner, M.A.; Buller, R.M.; Damon, I.K.; Lefkowitz, E.J.; McFadden, G.; McInnes, C.J.; Mercer, A.A.; Moyer, R.W.; Upton, C. Poxviridae. Available online: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/dsdna-viruses-2011/w/dsdna_viruses/74/poxviridae (accessed on 15 May 2019).
- Mauldin, M.R.; Antwerpen, M.; Emerson, G.L.; Li, Y.; Zoeller, G.; Carroll, D.S.; Meyer, H. Cowpox Virus: What’s in a Name? Viruses 2017, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D.S.; Emerson, G.L.; Li, Y.; Sammons, S.; Olson, V.; Frace, M.; Nakazawa, Y.; Czerny, C.P.; Tryland, M.; Kolodziejek, J.; et al. Chasing Jenner’s vaccine: Revisiting cowpox virus classification. PLoS ONE 2011, 6, e23086. [Google Scholar] [CrossRef]
- Smithson, C.; Tang, N.; Sammons, S.; Frace, M.; Batra, D.; Li, Y.; Emerson, G.L.; Carroll, D.S.; Upton, C. The genomes of three North American orthopoxviruses. Virus Genes 2017, 53, 21–34. [Google Scholar] [CrossRef]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer Nature: Basingstoke, UK, 2016. [Google Scholar]
- Bouckaert, R.; Heled, J.; Kuhnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef]
- Felsenstein, J. Distance Methods for Inferring Phylogenies: A Justification. Evolution 1984, 38, 16–24. [Google Scholar] [CrossRef]
- Wickham, H.; Henry, L. Tidyr: Easily Tidy Data with ‘Spread()’ and ‘Gather()’ Functions; R Package Version 0.8.0. 2018. Available online: https://CRAN.R-project.org/package=tidyr (accessed on 25 April 2019).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- RStudio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2016. [Google Scholar]
- Gubser, C.; Hue, S.; Kellam, P.; Smith, G.L. Poxvirus genomes: A phylogenetic analysis. J. Gen. Virol. 2004, 85, 105–117. [Google Scholar] [CrossRef]
- Fleischauer, C.; Upton, C.; Victoria, J.; Jones, G.J.; Roper, R.L. Genome sequence and comparative virulence of raccoonpox virus: The first North American poxvirus sequence. J. Gen. Virol. 2015, 96, 2806–2821. [Google Scholar] [CrossRef]
- Upton, C.; Slack, S.; Hunter, A.L.; Ehlers, A.; Roper, R.L. Poxvirus orthologous clusters: Toward defining the minimum essential poxvirus genome. J. Virol. 2003, 77, 7590–7600. [Google Scholar] [CrossRef]
- Lefkowitz, E.J.; Wang, C.; Upton, C. Poxviruses: Past, present and future. Virus Res. 2006, 117, 105–118. [Google Scholar] [CrossRef]
- Coulson, D.; Upton, C. Characterization of indels in poxvirus genomes. Virus Genes 2011, 42, 171–177. [Google Scholar] [CrossRef]
- Esposito, J.J.; Sammons, S.A.; Frace, A.M.; Osborne, J.D.; Olsen-Rasmussen, M.; Zhang, M.; Govil, D.; Damon, I.K.; Kline, R.; Laker, M.; et al. Genome sequence diversity and clues to the evolution of variola (smallpox) virus. Science 2006, 313, 807–812. [Google Scholar] [CrossRef]
- McLysaght, A.; Baldi, P.F.; Gaut, B.S. Extensive gene gain associated with adaptive evolution of poxviruses. Proc. Natl. Acad. Sci. USA 2003, 100, 15655–15660. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Droit, L.; Tesh, R.B.; Popov, V.L.; Little, N.S.; Upton, C.; Virgin, H.W.; Wang, D. The genome of Yoka poxvirus. J. Virol. 2011, 85, 10230–10238. [Google Scholar] [CrossRef]
- Price, N.; Tscharke, D.C.; Hollinshead, M.; Smith, G.L. Vaccinia virus gene B7R encodes an 18-kDa protein that is resident in the endoplasmic reticulum and affects virus virulence. Virology 2000, 267, 65–79. [Google Scholar] [CrossRef]
- Bratke, K.A.; McLysaght, A.; Rothenburg, S. A survey of host range genes in poxvirus genomes. Infect. Genet. Evol. 2013, 14, 406–425. [Google Scholar] [CrossRef]
- Hughes, A.L.; Friedman, R. Poxvirus genome evolution by gene gain and loss. Mol. Phylogenet. Evol. 2005, 35, 186–195. [Google Scholar] [CrossRef]
- Jarahian, M.; Fiedler, M.; Cohnen, A.; Djandji, D.; Hammerling, G.J.; Gati, C.; Cerwenka, A.; Turner, P.C.; Moyer, R.W.; Watzl, C.; et al. Modulation of NKp30- and NKp46-mediated natural killer cell responses by poxviral hemagglutinin. PLoS Pathog. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Buller, R.M.; Palumbo, G.J. Poxvirus pathogenesis. Microbiol. Rev. 1991, 55, 80–122. [Google Scholar]
- Fenner, F. Mousepox (infectious ectromelia): Past, present, and future. Lab. Anim. Sci. 1981, 31, 553–559. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CPXV | AKMV | Old World | New World | YPV | |
|---|---|---|---|---|---|
| AKPV | 92.93 | 93.52 | 92.87 | 87.10 | 74.88 |
| CPXV | 98.44 | 95.62 | 97.52 | 87.38 | 74.78 |
| AKMV | 99.39 | 95.48 | 87.27 | 74.72 | |
| Old World | 97.52 | 87.29 | 74.79 | ||
| New World | 92.02 | 75.58 |
| Amino Acid Identity | ||
|---|---|---|
| AKPV011 | AKPV013 | |
| SKPV | 93.00 | 90.74 |
| RCNV | 92.50 | 92.42 |
| CPXV | 90.00 | 88.50 |
| ECTV | 87.13 | 89.40 |
| AKMV | 86.57 | 80.05 |
| Length | AKPV | AKMV | OPVA | RCNV | |
|---|---|---|---|---|---|
| Region 1 | 7794 | 92.99 | 95.41 | 97.95 | 87.21 |
| Region 2 | 3882 | 94.50 | 76.11 | 67.55 | 64.35 |
| Region 3 | 11,816 | 86.50 | 91.34 | 95.59 | 75.15 |
| Region 4 | 1946 | 91.93 | 82.59 | 75.66 | 63.35 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gigante, C.M.; Gao, J.; Tang, S.; McCollum, A.M.; Wilkins, K.; Reynolds, M.G.; Davidson, W.; McLaughlin, J.; Olson, V.A.; Li, Y. Genome of Alaskapox Virus, a Novel Orthopoxvirus Isolated from Alaska. Viruses 2019, 11, 708. https://doi.org/10.3390/v11080708
Gigante CM, Gao J, Tang S, McCollum AM, Wilkins K, Reynolds MG, Davidson W, McLaughlin J, Olson VA, Li Y. Genome of Alaskapox Virus, a Novel Orthopoxvirus Isolated from Alaska. Viruses. 2019; 11(8):708. https://doi.org/10.3390/v11080708
Chicago/Turabian StyleGigante, Crystal M., Jinxin Gao, Shiyuyun Tang, Andrea M. McCollum, Kimberly Wilkins, Mary G. Reynolds, Whitni Davidson, Joseph McLaughlin, Victoria A. Olson, and Yu Li. 2019. "Genome of Alaskapox Virus, a Novel Orthopoxvirus Isolated from Alaska" Viruses 11, no. 8: 708. https://doi.org/10.3390/v11080708
APA StyleGigante, C. M., Gao, J., Tang, S., McCollum, A. M., Wilkins, K., Reynolds, M. G., Davidson, W., McLaughlin, J., Olson, V. A., & Li, Y. (2019). Genome of Alaskapox Virus, a Novel Orthopoxvirus Isolated from Alaska. Viruses, 11(8), 708. https://doi.org/10.3390/v11080708