Molecular Evolution of Classic Human Astrovirus, as Revealed by the Analysis of the Capsid Protein Gene

Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Genetic Diversity Analysis
2.3. Root-to-Tip Divergence Analysis
2.4. Accumulation Pattern of Amino Acid Substitutions
2.5. Evolutionary Analysis
2.6. Selection Pressure Analysis
3. Results
3.1. Description of Classic HAstV ORF2 Sequences in the GenBank Database
3.2. Genetic Diversity of Classic HAstV ORF2 Sequences
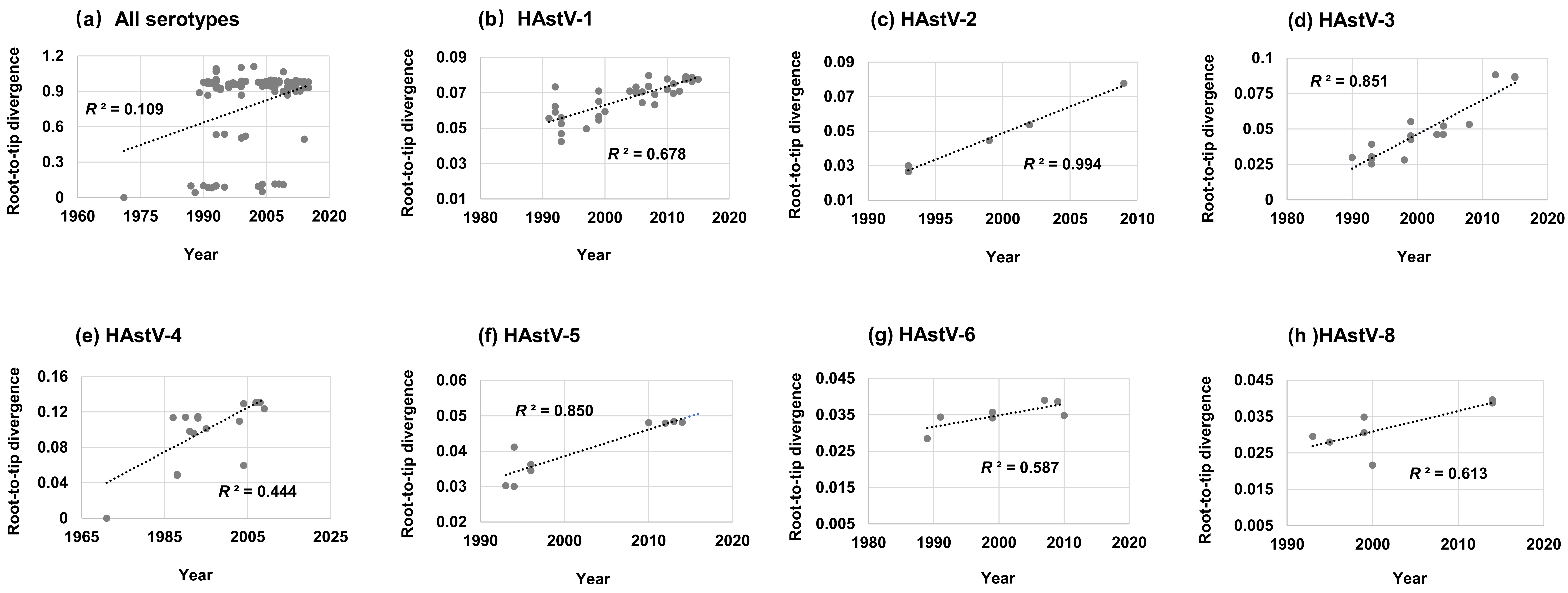
3.3. Root-to-Tip Divergence Analysis
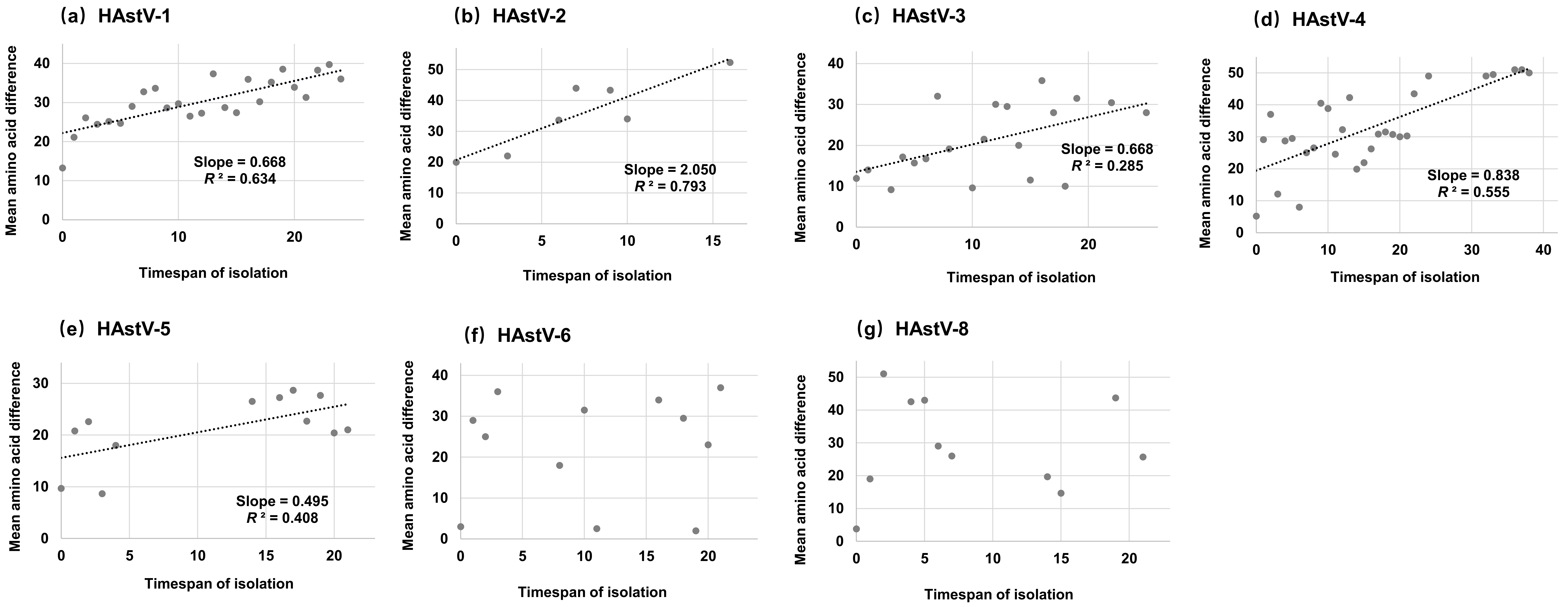
3.4. Accumulation Pattern of Amino Acid Substitutions
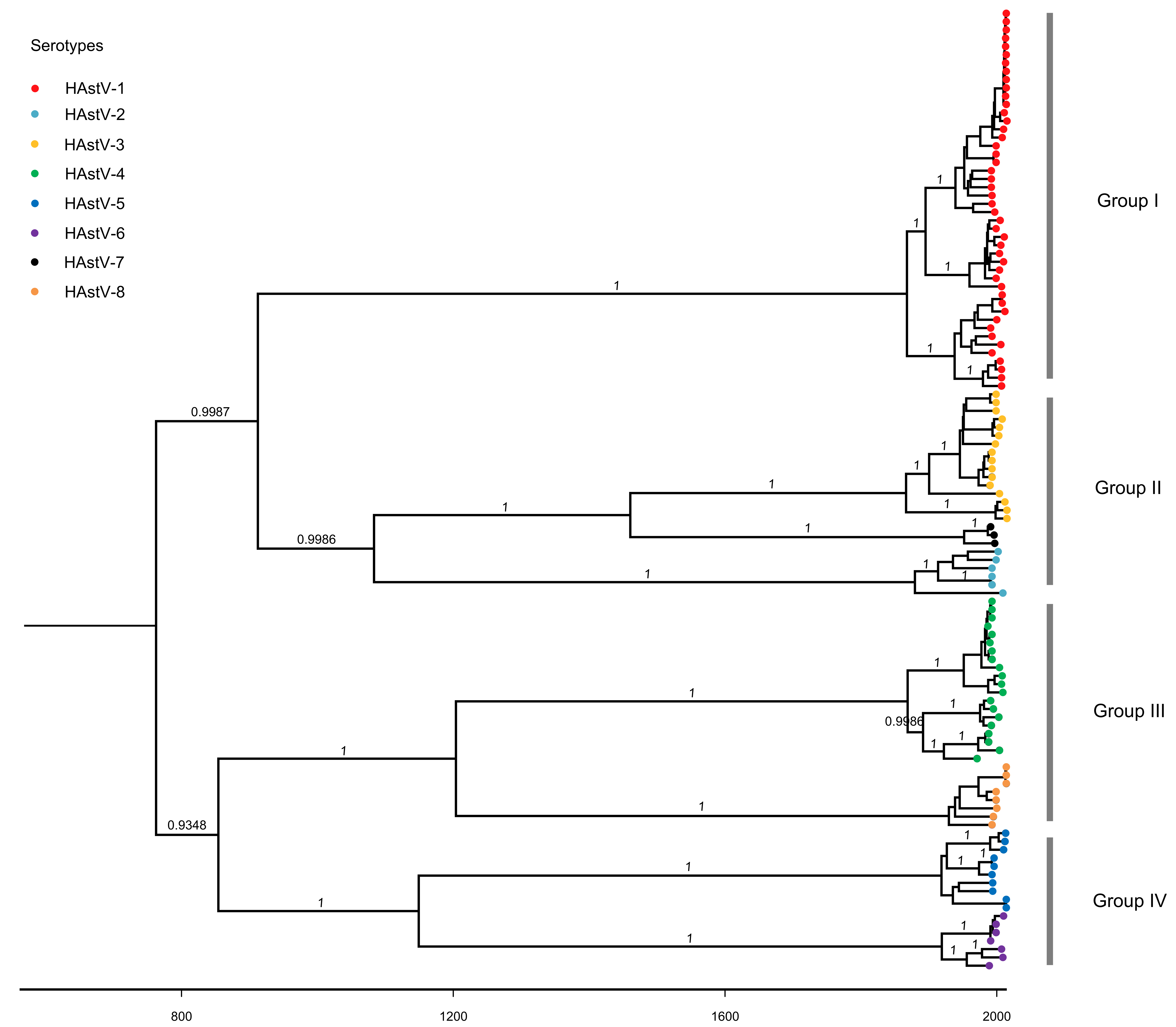
3.5. Time-Scale Phylogenetic Tree
3.6. Evolutionary Rate of ORF2 Sequences
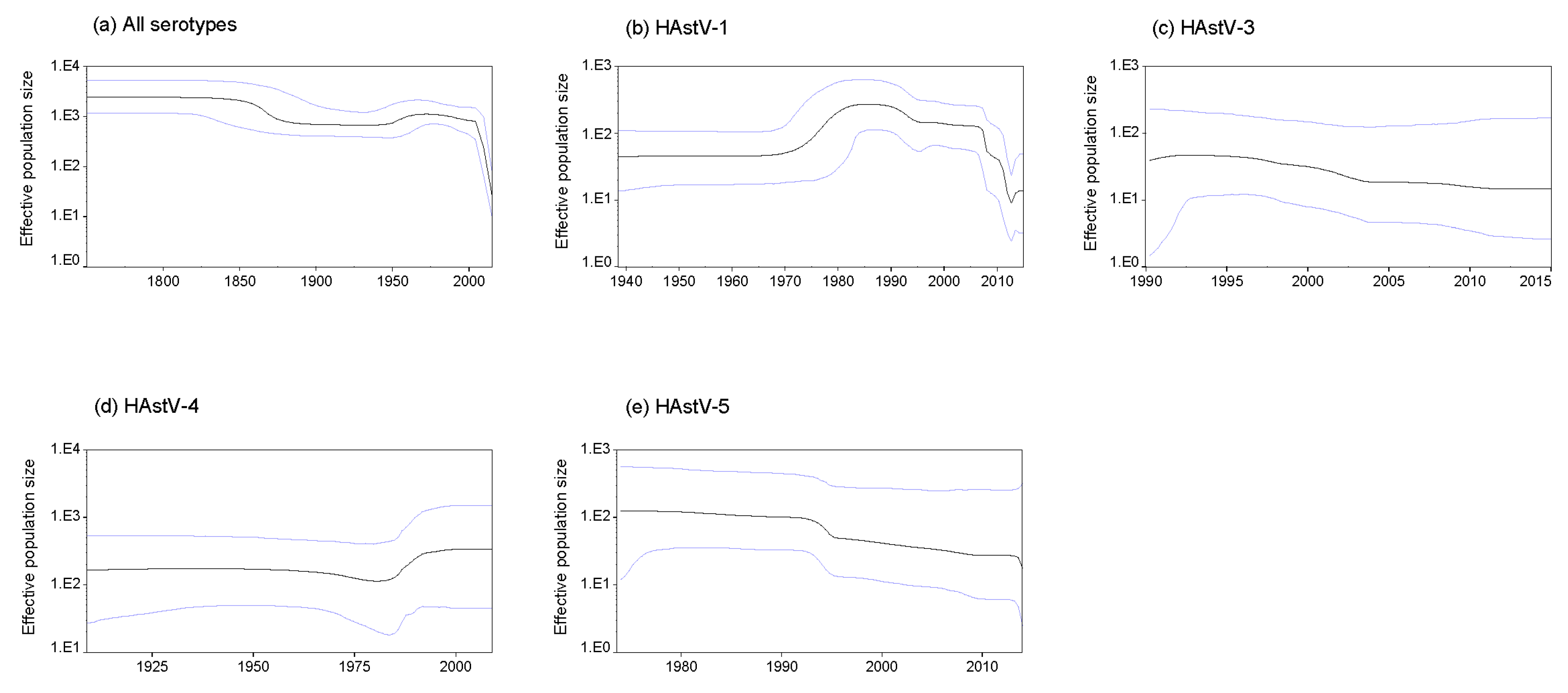
3.7. Phylodynamics of Classic HAstVs Strains
3.8. Selective Pressure Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Perot, P.; Lecuit, M.; Eloit, M. Astrovirus Diagnostics. Viruses 2017, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.F.; DuBois, R.M. The Astrovirus Capsid: A Review. Viruses 2017, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.; Pinto, R.M.; Guix, S. Human astroviruses. Clin. Microbiol. Rev. 2014, 27, 1048–1074. [Google Scholar] [CrossRef] [PubMed]
- De Benedictis, P.; Schultz-Cherry, S.; Burnham, A.; Cattoli, G. Astrovirus infections in humans and animals—Molecular biology, genetic diversity, and interspecies transmissions. Infect. Genet. Evol. 2011, 11, 1529–1544. [Google Scholar] [CrossRef]
- Appleton, H.; Higgins, P.G. Letter: Viruses and gastroenteritis in infants. Lancet 1975, 1, 1297. [Google Scholar] [CrossRef]
- Cortez, V.; Meliopoulos, V.A.; Karlsson, E.A.; Hargest, V.; Johnson, C.; Schultz-Cherry, S. Astrovirus biology and pathogenesis. Annu. Rev. Virol. 2017, 4, 327–348. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Li, L.; Victoria, J.; Oderinde, B.; Mason, C.; Pandey, P.; Zaidi, S.Z.; Delwart, E. Multiple novel astrovirus species in human stool. J. Gen. Virol. 2009, 90, 2965–2972. [Google Scholar] [CrossRef]
- Kumthip, K.; Khamrin, P.; Ushijima, H.; Maneekarn, N. Molecular epidemiology of classic, MLB and VA astroviruses isolated from <5 year-old children with gastroenteritis in Thailand, 2011–2016. Infect. Genet. Evol. 2018, 65, 373–379. [Google Scholar]
- Tao, Z.; Wang, H.; Zhang, W.; Xu, A. Novel astrovirus types circulating in Shandong Province (Eastern China) during 2016: A clinical and environmental surveillance. J. Clin. Virol. 2019, 116, 69–73. [Google Scholar] [CrossRef]
- Vu, D.L.; Cordey, S.; Brito, F.; Kaiser, L. Novel human astroviruses: Novel human diseases? J. Clin. Virol. 2016, 82, 56–63. [Google Scholar] [CrossRef]
- Méndez, E.; Arias, C. Astroviruses. In Fields Virology, 6th ed.; Knipe, D., Howley, P., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Lyman, W.H.; Walsh, J.F.; Kotch, J.B.; Weber, D.J.; Gunn, E.; Vinjé, J. Prospective study of etiologic agents of acute gastroenteritis outbreaks in child care centers. J. Pediatr. 2006, 154, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Matsushima, Y.; Nagasawa, K.; Motoya, T.; Ryo, A.; Kuroda, M.; Katayama, K.; Kimura, H. Molecular evolutionary analyses of the RNA-dependent RNA polymerase region in norovirus genogroup II. Front. Microbiol. 2018, 9, 3070. [Google Scholar] [CrossRef] [PubMed]
- Zeller, M.; Heylen, E.; Damanka, S.; Pietsch, C.; Donato, C.; Tamura, T.; Kulkarni, R.; Arora, R.; Cunliffe, N.; Maunula, L.; et al. Emerging OP354-Like P [8] rotaviruses have rapidly dispersed from Asia to other continents. Mol. Biol. Evol. 2015, 32, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Shi, W.; Shi, Y.; Wang, D.; Xiao, H.; Li, W.; Bi, Y.; Wu, Y.; Li, X.; Yan, J.; et al. Origin and diversity of novel avian influenza A H7N9 viruses causing human infection: Phylogenetic, structural, and coalescent analyses. Lancet 2013, 381, 1926–1932. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit_a user-friendly biological sequence alignment editor and analysis program for Windows 95_98_NT. Nucleic Acids Res. Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Suchard, M.A.; Weiss, R.E.; Sinsheimer, J.S. Bayesian selection of continuous-time markov chain evolutionary models. Mol. Biol. Evol. 2001, 18, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Tohma, K.; Lepore, C.J.; Ford-Siltz, L.A.; Parra, G.I. Phylogenetic analyses suggest that factors other than the capsid protein play a role in the epidemic potential of GII.2 norovirus. mSphere 2017, 2, e00187-17. [Google Scholar] [CrossRef] [PubMed]
- Tohma, K.; Lepore, C.J.; Ford-Siltz, L.A.; Parra, G.I. Evolutionary dynamics of non-GII genotype 4 (GII.4) noroviruses reveal limited and independent diversification of variants. J. Gen. Virol. 2018, 99, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Martella, V.; Pinto, P.; Tummolo, F.; De Grazia, S.; Giammanco, G.M.; Medici, M.C.; Ganesh, B.; L’Homme, Y.; Farkas, T.; Jakab, F.; et al. Analysis of the ORF2 of human astroviruses reveals lineage diversification, recombination and rearrangement and provides the basis for a novel sub-classification system. Arch. Virol. 2014, 159, 3185–3196. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef]
- Lauring, A.S. Complexities of viral mutation rates. J. Virol. 2018, 92, e01031-17. [Google Scholar]
- Wohlgemuth, N.; Honce, R.; Schultz-Cherry, S. Astrovirus evolution and emergence. Infect. Genet. Evol. 2019, 69, 30–37. [Google Scholar] [CrossRef]
- Babkin, I.V.; Tikunov, A.Y.; Sedelnikova, D.A.; Zhirakovskaia, E.V.; Tikunova, N.V. Recombination analysis based on the HAstV-2 and HAstV-4 complete genomes. Infect. Genet. Evol. 2014, 22, 94–102. [Google Scholar] [CrossRef]
- De Grazia, S.; Medici, M.C.; Pinto, P.; Moschidou, P.; Tummolo, F.; Calderaro, A.; Bonura, F.; Banyai, K.; Giammanco, G.M.; Martella, V. Genetic heterogeneity and recombination in human type 2 astroviruses. J. Clin. Microbiol. 2012, 50, 3760–3764. [Google Scholar] [CrossRef] [PubMed]
- Medici, M.C.; Tummolo, F.; Martella, V.; Banyai, K.; Bonerba, E.; Chezzi, C.; Arcangeletti, M.C.; De Conto, F.; Calderaro, A. Genetic heterogeneity and recombination in type-3 human astroviruses. Infect. Genet. Evol. 2015, 32, 156–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra, G.I.; Squires, R.B.; Karangwa, C.K.; Johnson, J.A.; Lepore, C.J.; Sosnovtsev, S.V.; Green, K.Y. Static and evolving norovirus genotypes: Implications for epidemiology and immunity. PLoS Pathog. 2017, 13, e1006136. [Google Scholar] [CrossRef] [PubMed]
- Vu, D.L.; Bosch, A.; Pinto, R.M.; Guix, S. Epidemiology of classic and novel human astrovirus: Gastroenteritis and beyond. Viruses 2017, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Belliot, G.; Laveran, H.; Monroe, S.S. Detection and genetic differentiation of human astroviruses_ phylogenetic grouping varies by coding region. Arch. Virol. 1997, 142, 1323–1334. [Google Scholar] [CrossRef]
- Guix, S.; Caballero, S.; Fuentes, C.; Bosch, A.; Pinto, R.M. Genetic analysis of the hypervariable region of the human astrovirus nsP1a coding region: Design of a new RFLP typing method. J. Med. Virol. 2008, 80, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Cobey, S.; Koelle, K. Capturing escape in infectious disease dynamics. Trends Ecol. Evol. 2008, 23, 572–577. [Google Scholar] [CrossRef]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Hanada, K.; Suzuki, Y.; Gojobori, T. A large variation in the rates of synonymous substitution for RNA viruses and its relationship to a diversity of viral infection and transmission modes. Mol. Biol. Evol. 2004, 21, 1074–1080. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Rambaut, A.; Pybus, O.G.; Holmes, E.C. Rates of molecular evolution in RNA viruses: A quantitative phylogenetic analysis. J. Mol. Evol. 2002, 54, 156–165. [Google Scholar] [CrossRef]
- Babkin, I.V.; Tikunov, A.Y.; Zhirakovskaia, E.V.; Netesov, S.V.; Tikunova, N.V. High evolutionary rate of human astrovirus. Infect. Genet. Evol. 2012, 12, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Matsushima, Y.; Motoya, T.; Sakon, N.; Shigemoto, N.; Okamoto-Nakagawa, R.; Nishimura, K.; Yamashita, Y.; Kuroda, M.; Saruki, N.; et al. Molecular evolution of the capsid gene in human norovirus genogroup II. Sci. Rep. 2016, 6, 29400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, M.; Yoshizumi, S.; Kogawa, S.; Takahashi, T.; Ueki, Y.; Shinohara, M.; Mizukoshi, F.; Tsukagoshi, H.; Sasaki, Y.; Suzuki, R.; et al. Molecular evolution of the capsid gene in norovirus genogroup I. Sci. Rep. 2015, 5, 13806. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Mahar, J.E.; Bok, K.; Green, K.Y.; Kirkwood, C.D. The importance of intergenic recombination in norovirus GII.3 evolution. J. Virol. 2013, 87, 3687–3698. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Strain, E.; Kelley, L.A.; Schultz-Cherry, S.; Muse, S.V.; Koci, M.D. Genomic analysis of closely related astroviruses. J. Virol. 2008, 82, 5099–5103. [Google Scholar] [CrossRef]
- Van Hemert, F.J.; Lukashov, V.V.; Berkhout, B. Different rates of (non-)synonymous mutations in astrovirus genes; correlation with gene function. Virol. J. 2007, 4, 25. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serotype | No. of Sequences | Years | Duration of Collection Years | Similarity | Inter-Serotype Mean Amino Acid Distance | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide | Amino Acid | HAstV-1 | HAstV-2 | HAstV-3 | HAstV-4 | HAstV-5 | HAstV-6 | HAstV-7 | ||||
| HAstV-1 | 46 | 1991–2015 | 24 | 89.4–100% | 90.9–100% | |||||||
| HAstV-2 | 6 | 1993–2009 | 16 | 87.6–99.8% | 88.7–99.8% | 0.341 | ||||||
| HAstV-3 | 16 | 1990–2015 | 25 | 88.2–99.7% | 92.9–99.8% | 0.242 | 0.305 | |||||
| HAstV-4 | 20 | 1971–2009 | 38 | 89.1–99.9% | 91.6–100% | 0.429 | 0.394 | 0.395 | ||||
| HAstV-5 | 10 | 1993–2014 | 21 | 93.1–100% | 96.0–100% | 0.327 | 0.389 | 0.305 | 0.410 | |||
| HAstV-6 | 7 | 1989–2010 | 21 | 93.5–99.8% | 95.2–99.8% | 0.304 | 0.366 | 0.289 | 0.393 | 0.254 | ||
| HAstV-7 | 3 | 1991–1997 | 16 | 96.6–99.5% | 96.4–99.3% | 0.282 | 0.332 | 0.167 | 0.431 | 0.302 | 0.288 | |
| HAstV-8 | 8 | 1993–2014 | 21 | 93.6–100% | 91.5–100% | 0.320 | 0.333 | 0.296 | 0.314 | 0.293 | 0.299 | 0.329 |
| All | 116 | 1971–2015 | 44 | 58.8–100% | 57.5–100% | |||||||
| Serotypes | Substitution Rate (Substitutions/Site/Year) | Ratio of Rate (Codon 3/Codon 1 + 2) | ||
|---|---|---|---|---|
| Mean | 95% HPDs a | Mean | 95% HPDs | |
| HAstV-1 | 7.898 × 10−4 | 6.143 × 10−4–9.747 × 10−4 | 2.684 | 2.198–3.396 |
| HAstV-3 | 2.195 × 10−3 | 7.439 × 10−4–3.565 × 10−3 | 4.484 | 3.374–6.225 |
| HAstV-4 | 3.964 × 10−4 | 1.655 × 10−4–6.014 × 10−4 | 2.769 | 2.109–3.788 |
| HAstV-5 | 7.577 × 10−4 | 2.836 × 10−4–1.277 × 10−3 | 3.724 | 2.695–5.485 |
| All | 4.509 × 10−4 | 3.558 × 10−4–5.512 × 10−4 | 2.727 | 2.414–3.082 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, N.; Zhou, L.; Wang, B. Molecular Evolution of Classic Human Astrovirus, as Revealed by the Analysis of the Capsid Protein Gene. Viruses 2019, 11, 707. https://doi.org/10.3390/v11080707
Zhou N, Zhou L, Wang B. Molecular Evolution of Classic Human Astrovirus, as Revealed by the Analysis of the Capsid Protein Gene. Viruses. 2019; 11(8):707. https://doi.org/10.3390/v11080707
Chicago/Turabian StyleZhou, Nan, Lu Zhou, and Bei Wang. 2019. "Molecular Evolution of Classic Human Astrovirus, as Revealed by the Analysis of the Capsid Protein Gene" Viruses 11, no. 8: 707. https://doi.org/10.3390/v11080707
APA StyleZhou, N., Zhou, L., & Wang, B. (2019). Molecular Evolution of Classic Human Astrovirus, as Revealed by the Analysis of the Capsid Protein Gene. Viruses, 11(8), 707. https://doi.org/10.3390/v11080707