Herpes Simplex Virus Type 1–Encoded miR-H2-3p Manipulates Cytosolic DNA–Stimulated Antiviral Innate Immune Response by Targeting DDX41

Abstract
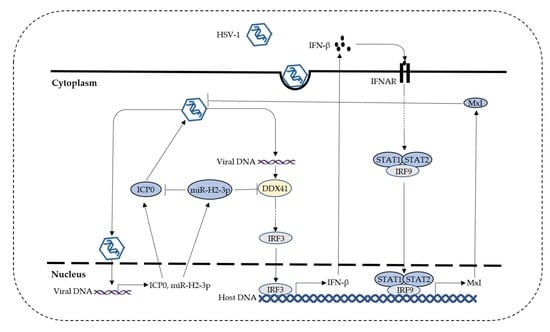
:
1. Introduction
2. Materials and Methods
2.1. Cells and Cell Culture
2.2. Virus and Viral Titer Assay
2.3. Transfection and Infection Assay
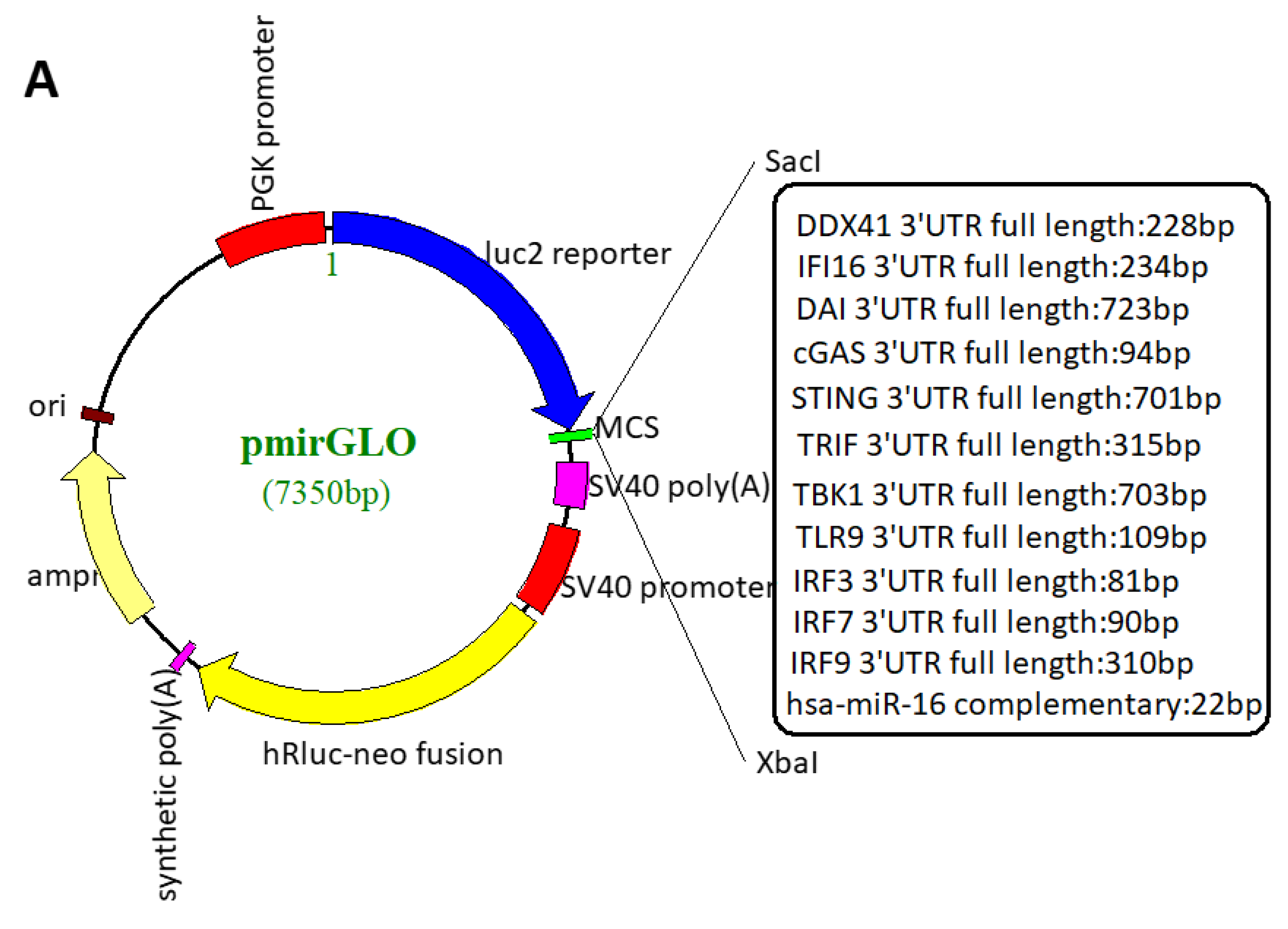
2.4. Plasmid Construction
2.5. Site-Directed Mutagenesis
2.6. Dual-Luciferase Reporter Assay
2.7. RNA Interference
2.8. miRNA Mimics and Inhibitors
2.9. RNA Quantification
2.10. QPCR Detection of Viral Load
2.11. Immunoblotting
2.12. Bioinformatics Analysis
2.13. Statistics and Data Analysis
3. Results
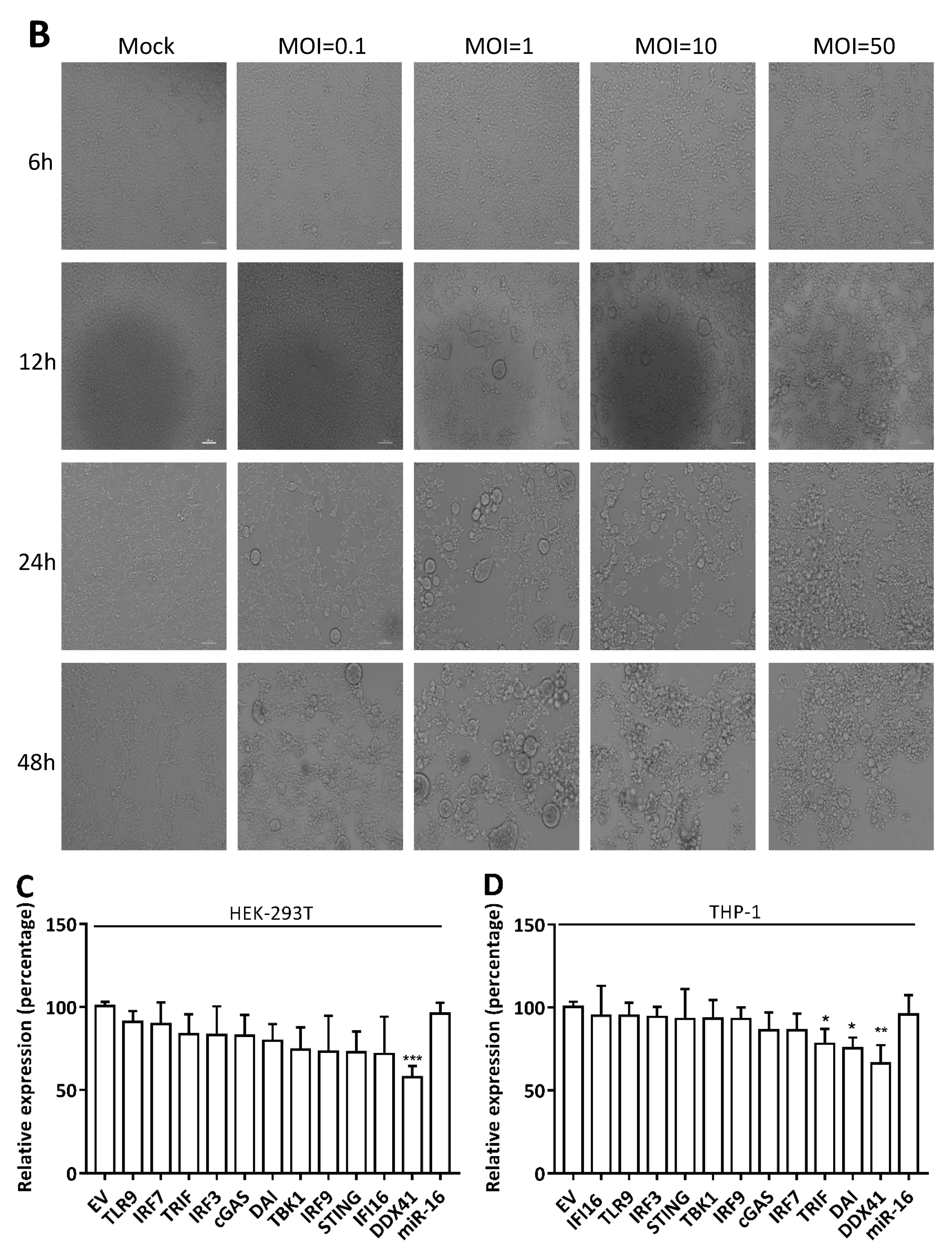
3.1. HSV-1 Infection Attenuated the Cytosolic DNA Sensor Reporter Expression of DDX41
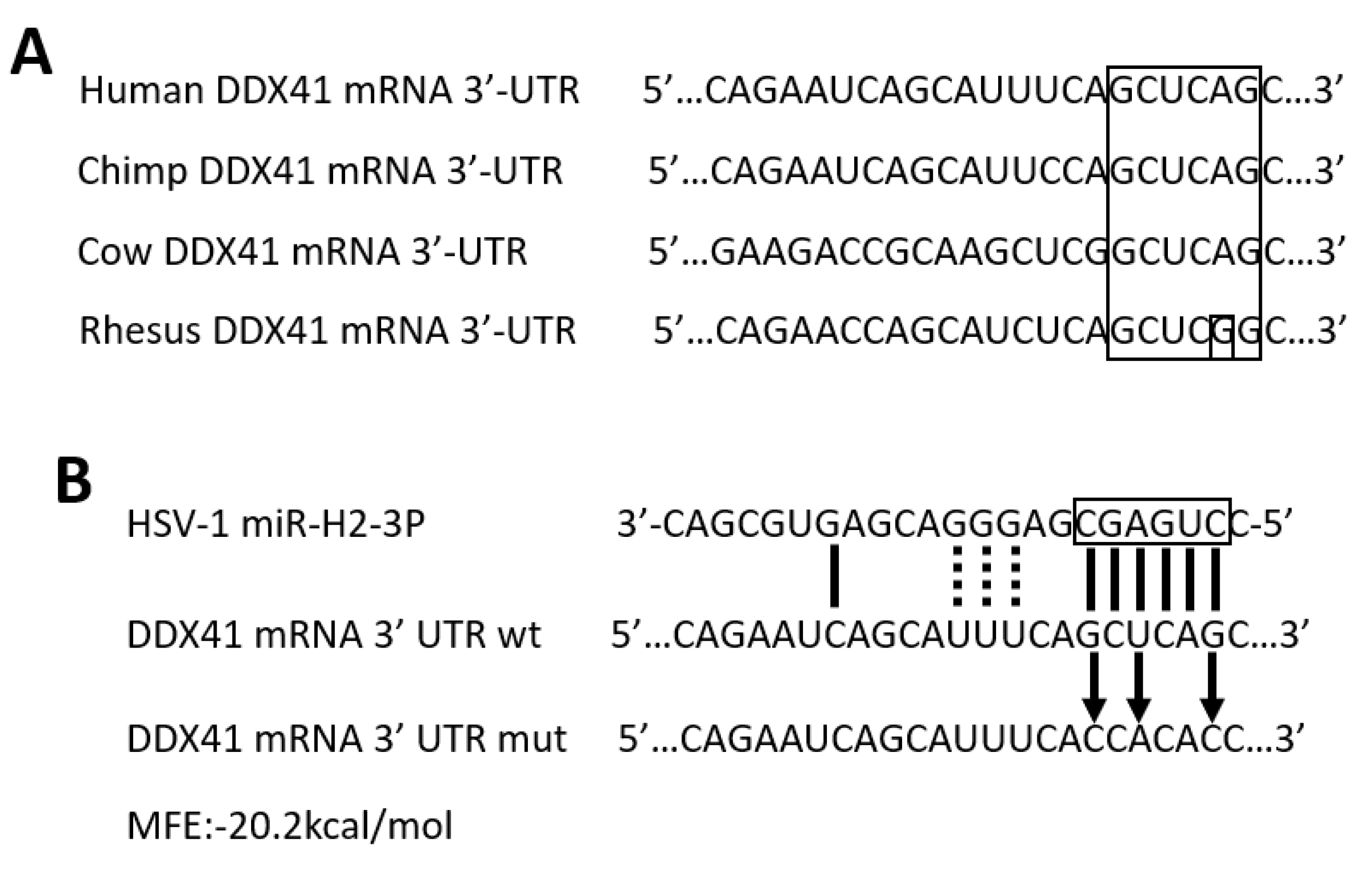
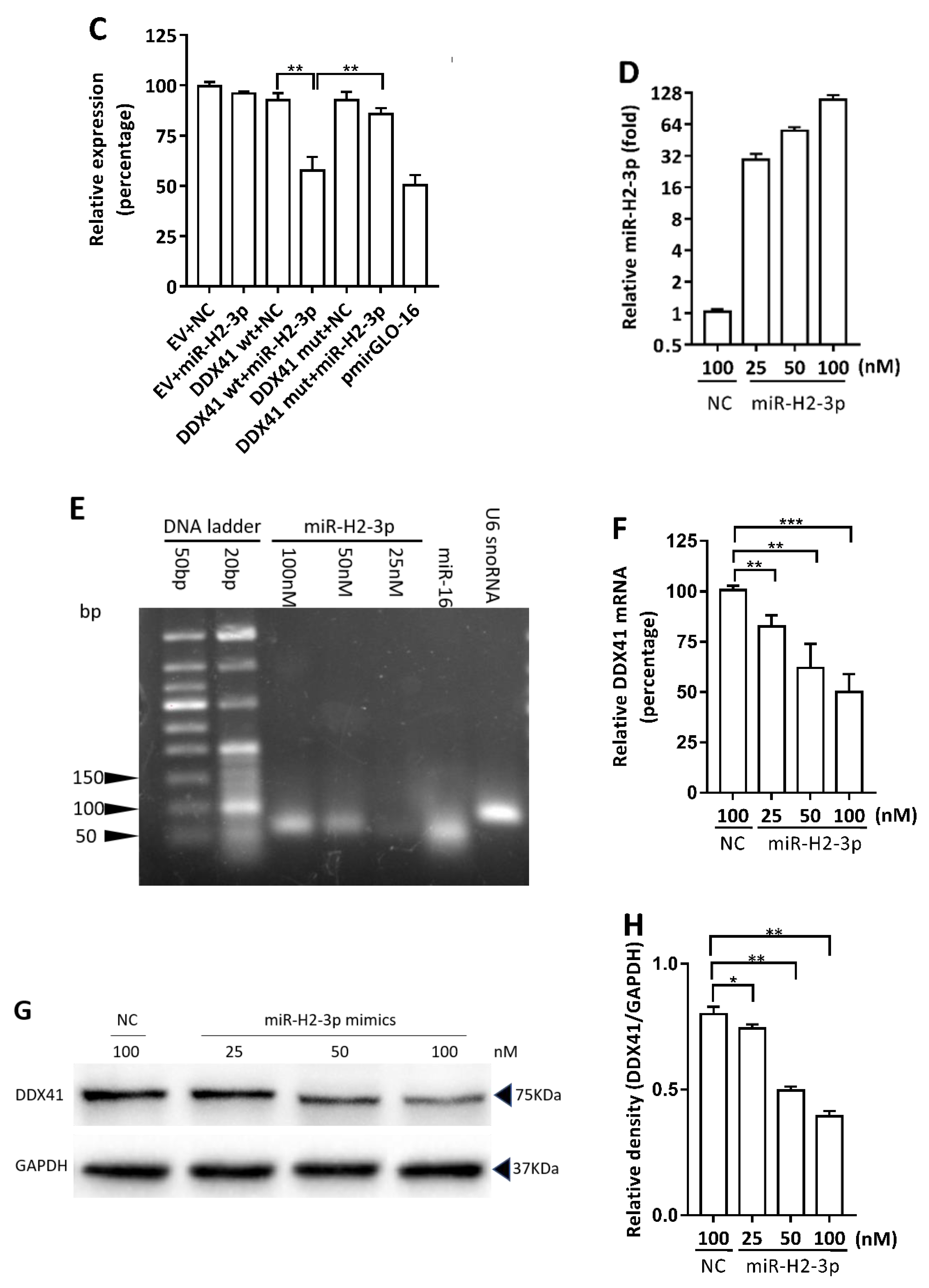
3.2. HSV-1-Encoded miR-H2-3p Targeted DDX41 Directly
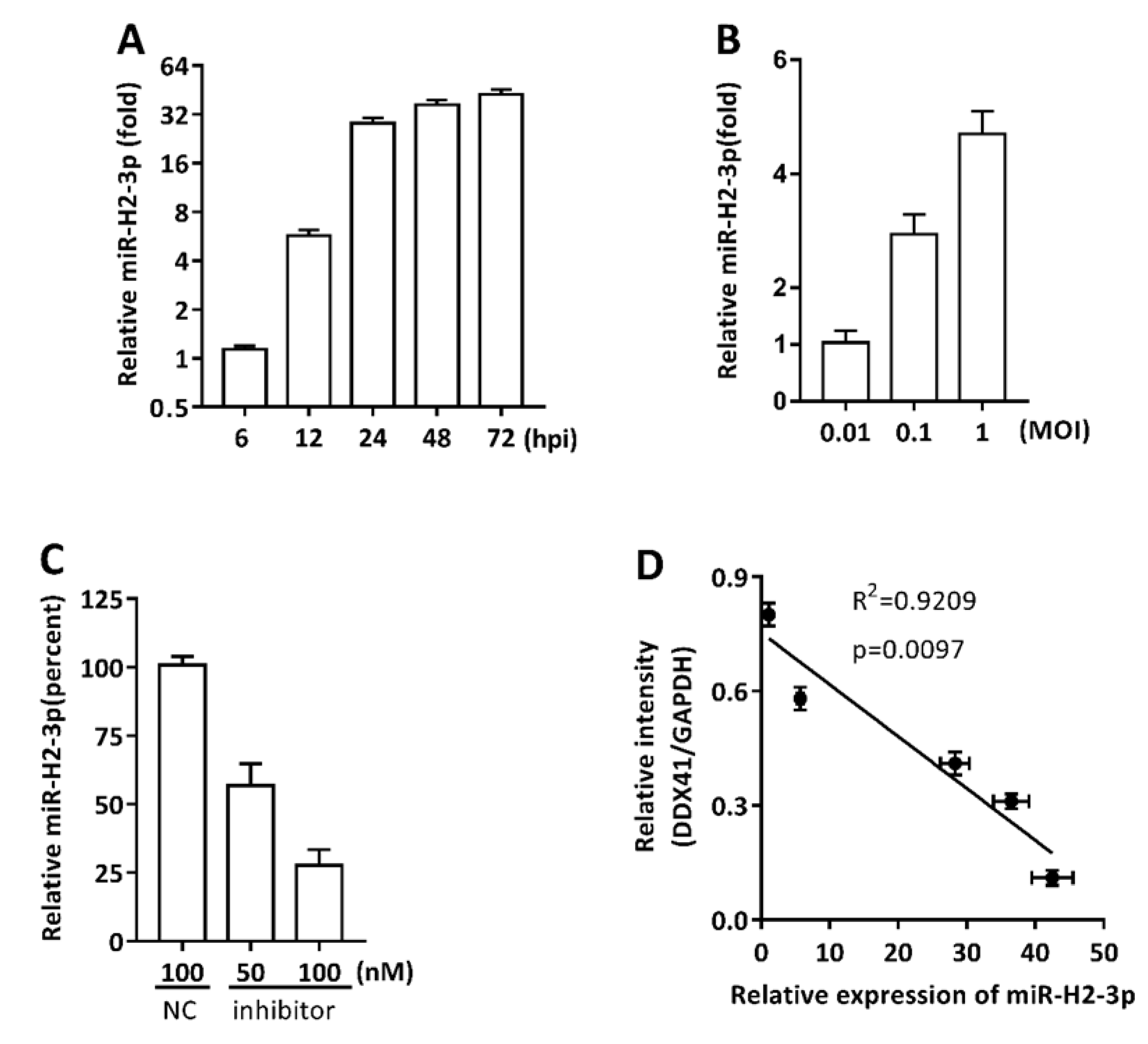
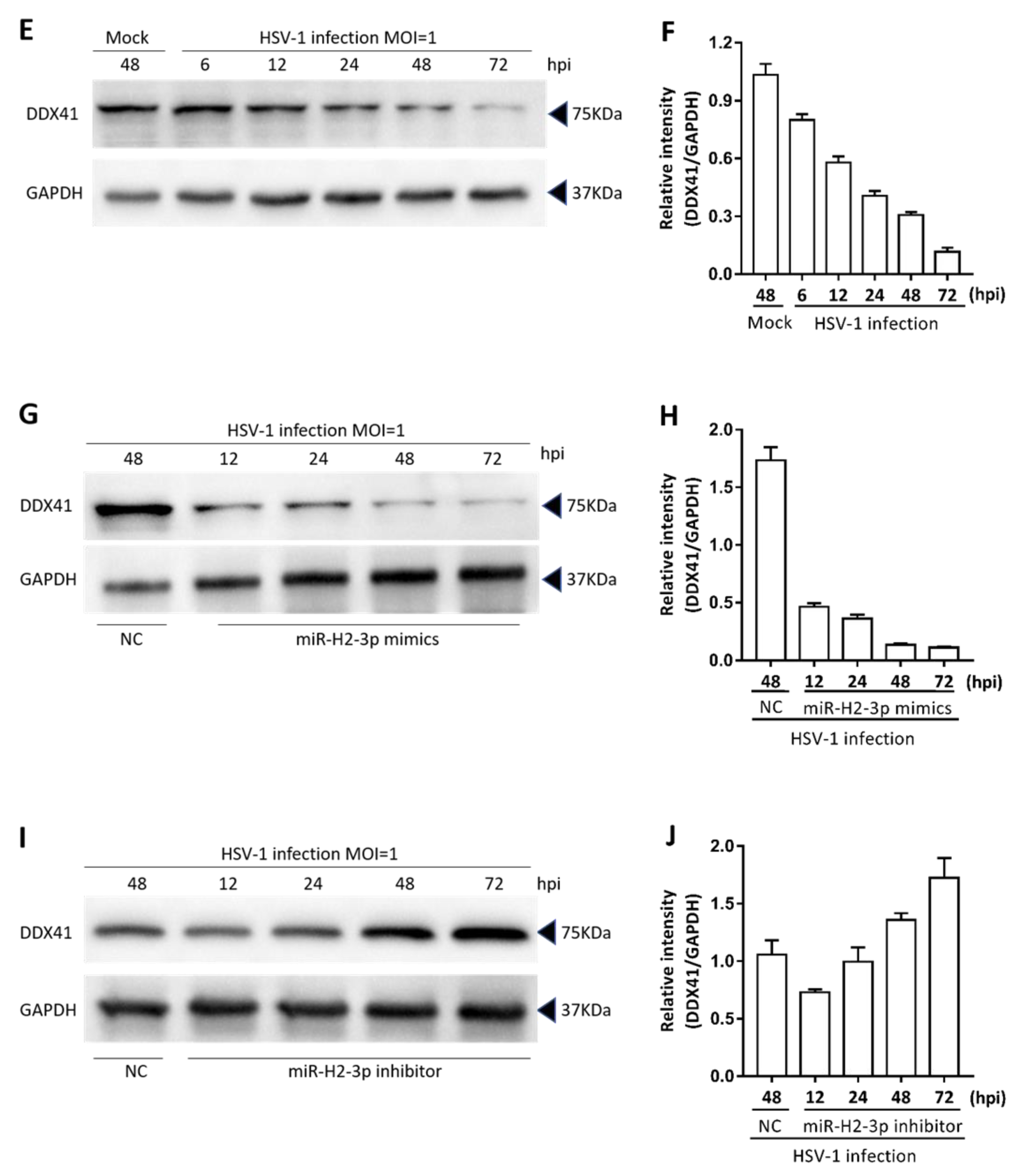
3.3. miR-H2-3p Contributed to the Downregulation of DDX41 during HSV-1 Infection in THP-1 Cells
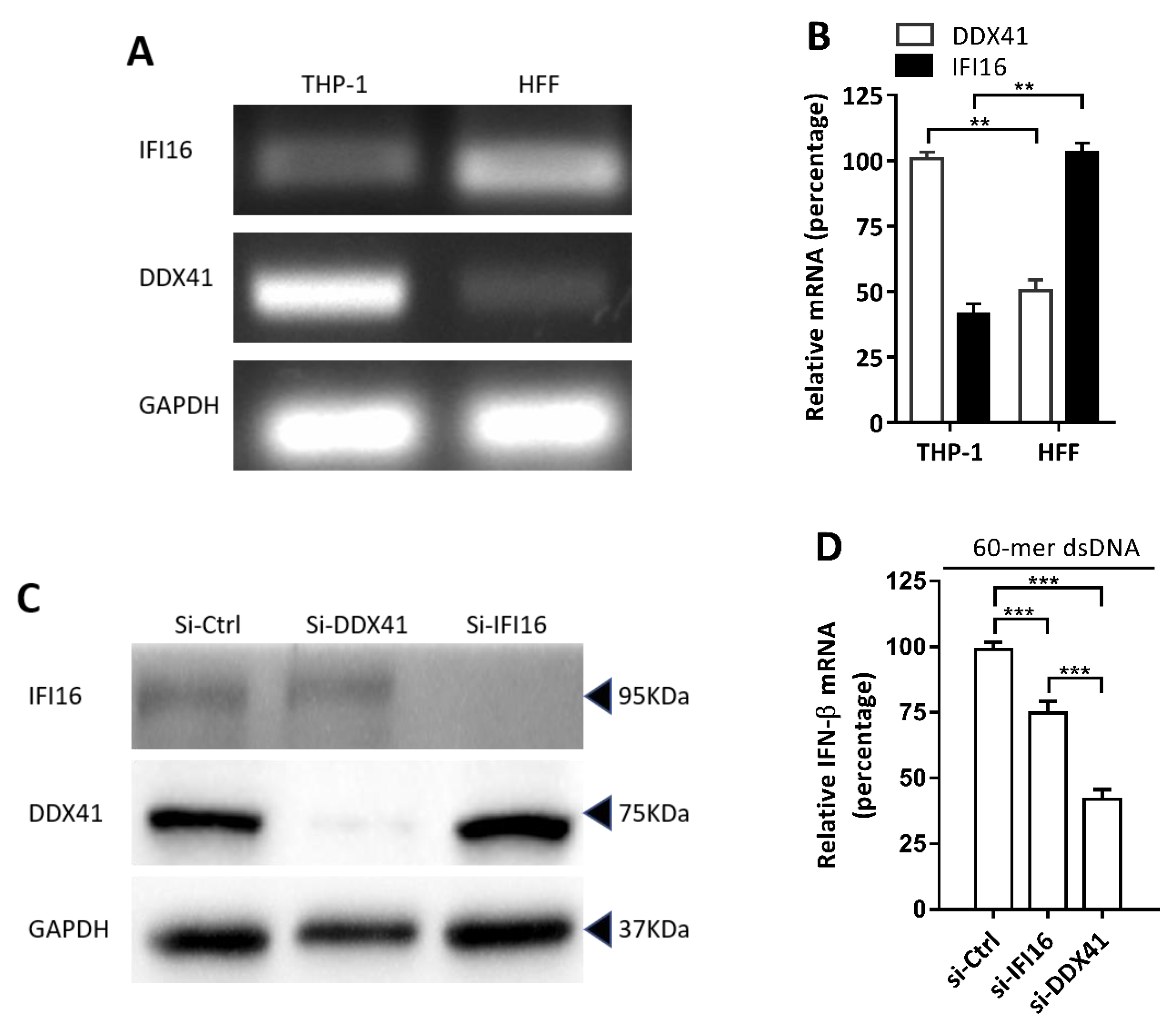
3.4. DDX41 was a Crucial DNA Sensor in THP-1 Cells
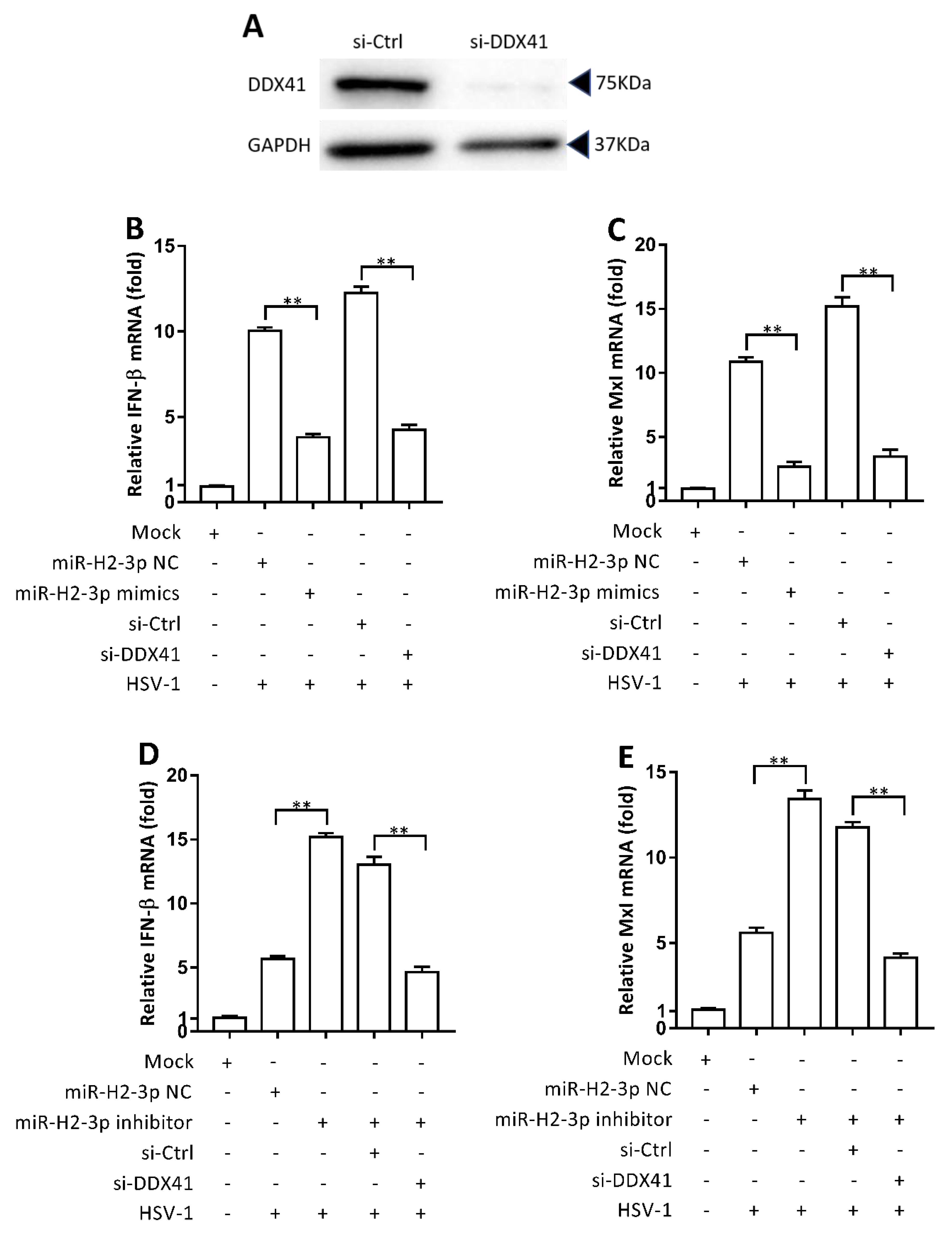
3.5. miR-H2-3p Negatively Regulated HSV-1-Triggered IFN-β and ISG Expression by Targeting DDX41
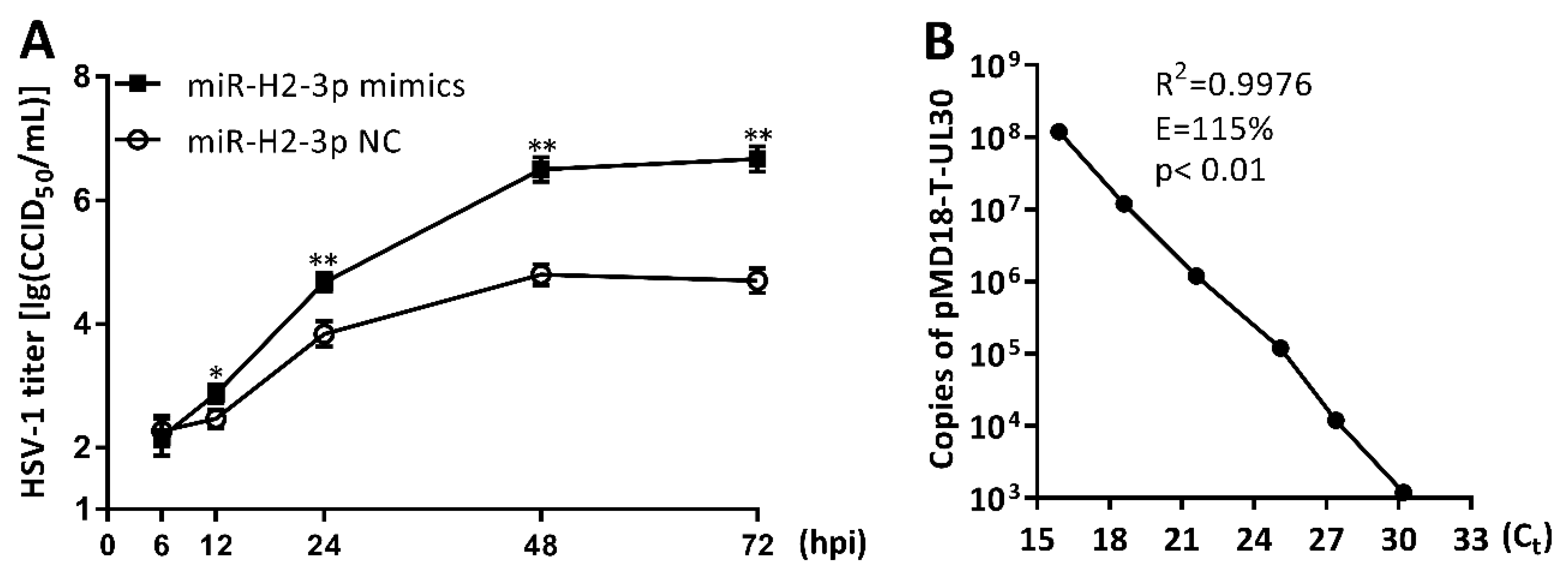
3.6. miR-H2-3p Promoted HSV-1 Replication and Viral mRNA Expression in THP-1 Cells
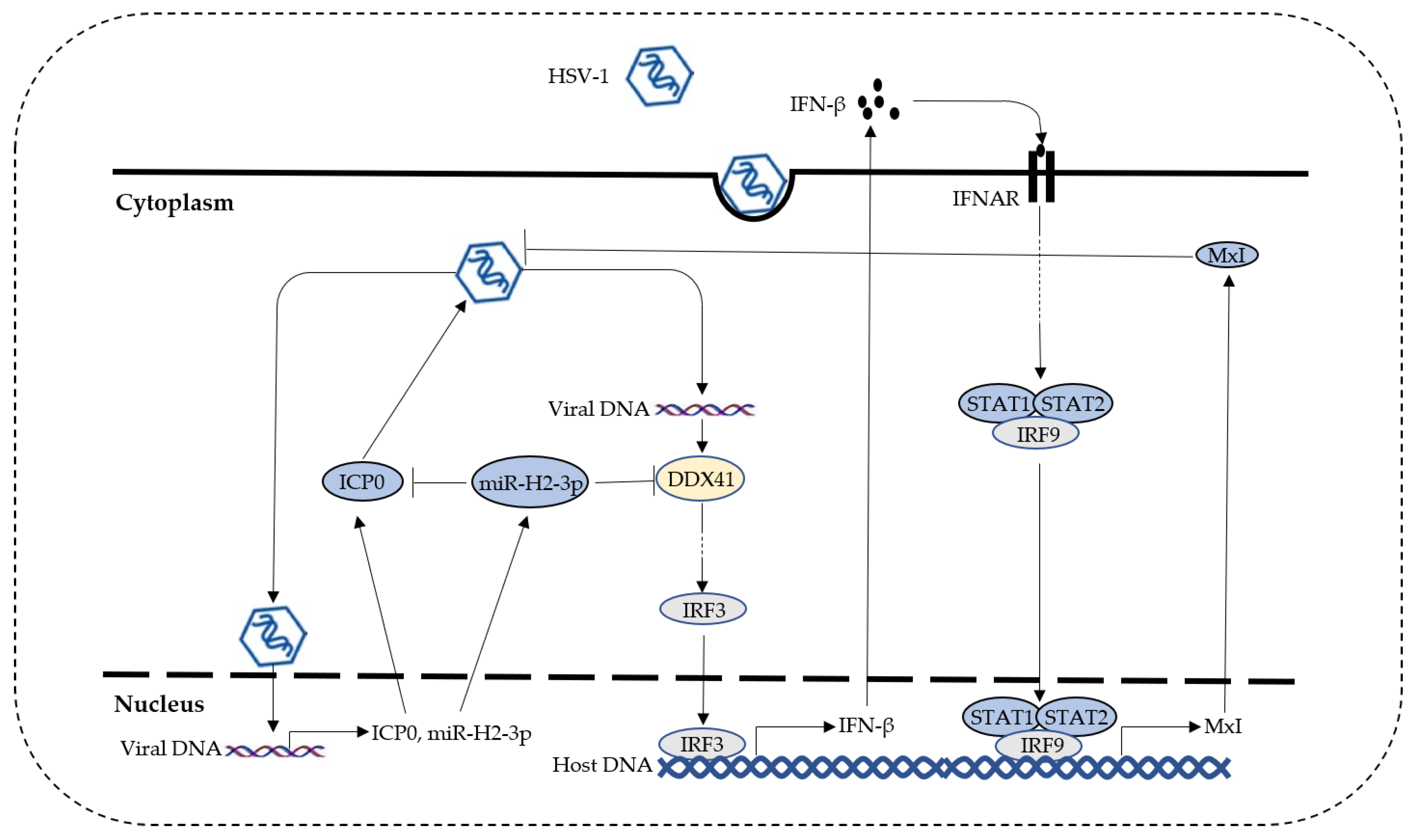
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. Dai (dlm-1/zbp1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. Ifi16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic gmp-amp synthase is a cytosolic DNA sensor that activates the type i interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.-J. The helicase ddx41 senses intracellular DNA mediated by the adaptor sting in dendritic cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Burckstummer, T.; Baumann, C.; Bluml, S.; Dixit, E.; Durnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; et al. An orthogonal proteomic-genomic screen identifies aim2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009, 10, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef] [PubMed]
- Jurak, I.; Silverstein, L.B.; Sharma, M.; Coen, D.M. Herpes simplex virus is equipped with rna- and protein-based mechanisms to repress expression of atrx, an effector of intrinsic immunity. J. Virol. 2012, 86, 10093–10102. [Google Scholar] [CrossRef] [PubMed]
- van Lint, A.L.; Murawski, M.R.; Goodbody, R.E.; Severa, M.; Fitzgerald, K.A.; Finberg, R.W.; Knipe, D.M.; Kurt-Jones, E.A. Herpes simplex virus immediate-early icp0 protein inhibits toll-like receptor 2-dependent inflammatory responses and nf-kappab signaling. J. Virol. 2010, 84, 10802–10811. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, K.; Wang, S.; Zheng, C. Herpes simplex virus 1 e3 ubiquitin ligase icp0 protein inhibits tumor necrosis factor alpha-induced nf-kappab activation by interacting with p65/rela and p50/nf-kappab1. J. Virol. 2013, 87, 12935–12948. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Chikoti, L.; Chandran, B. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the ifi16 and nlrp3 inflammasomes. J. Virol. 2013, 87, 5005–5018. [Google Scholar] [CrossRef] [PubMed]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear ifi16 induction of irf-3 signaling during herpesviral infection and degradation of ifi16 by the viral icp0 protein. Proc. Natl Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef]
- Cuchet-Lourenco, D.; Anderson, G.; Sloan, E.; Orr, A.; Everett, R.D. The viral ubiquitin ligase icp0 is neither sufficient nor necessary for degradation of the cellular DNA sensor ifi16 during herpes simplex virus 1 infection. J. Virol. 2013, 87, 13422–13432. [Google Scholar] [CrossRef] [PubMed]
- Kalamvoki, M.; Roizman, B. Hsv-1 degrades, stabilizes, requires, or is stung by sting depending on icp0, the us3 protein kinase, and cell derivation. Proc. Natl Acad. Sci. USA 2014, 111, E611–E617. [Google Scholar] [CrossRef] [PubMed]
- Lanfranca, M.P.; Mostafa, H.H.; Davido, D.J. Hsv-1 icp0: An e3 ubiquitin ligase that counteracts host intrinsic and innate immunity. Cell 2014, 3, 438–454. [Google Scholar] [CrossRef]
- Taddeo, B.; Roizman, B. The virion host shutoff protein (ul41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of rnase a. J. Virol. 2006, 80, 9341–9345. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Wang, K.; Wang, S.; Cai, M.; Li, M.l.; Zheng, C. Herpes simplex virus 1 counteracts viperin via its virion host shutoff protein ul41. J. Virol. 2014, 88, 12163–12166. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Su, C.; Zheng, C. Herpes simplex virus 1 tegument protein ul41 counteracts ifit3 antiviral innate immunity. J. Virol. 2016, 90, 11056–11061. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Zhang, J.; Zheng, C. Herpes simplex virus 1 ul41 protein abrogates the antiviral activity of hzap by degrading its mrna. Virol. J. 2015, 12, 203. [Google Scholar] [CrossRef] [PubMed]
- Zenner, H.L.; Mauricio, R.; Banting, G.; Crump, C.M. Herpes simplex virus 1 counteracts tetherin restriction via its virion host shutoff activity. J. Virol. 2013, 87, 13115–13123. [Google Scholar] [CrossRef]
- Sun, C.; Schattgen, S.A.; Pisitkun, P.; Jorgensen, J.P.; Hilterbrand, A.T.; Wang, L.J.; West, J.A.; Hansen, K.; Horan, K.A.; Jakobsen, M.R.; et al. Evasion of innate cytosolic DNA sensing by a gammaherpesvirus facilitates establishment of latent infection. J. Immunol. 2015, 194, 1819–1831. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Su, C.; Xu, H.; Zheng, C. Herpes simplex virus 1 ubiquitin-specific protease ul36 abrogates nf-kappab activation in DNA sensing signal pathway. J. Virol. 2017, 91, e02417-16. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; You, J.; You, H.; Zheng, C. Herpes simplex virus 1 ul36usp antagonizes type i interferon-mediated antiviral innate immunity. J. Virol. 2018, 92, e01161-18. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, K.; Li, J.; Zheng, C. Herpes simplex virus 1 ubiquitin-specific protease ul36 inhibits beta interferon production by deubiquitinating traf3. J. Virol. 2013, 87, 11851–11860. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.; Mohr, I. Inhibition of cellular 2′-5′ oligoadenylate synthetase by the herpes simplex virus type 1 us11 protein. J. Virol. 2007, 81, 3455–3464. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Wang, S.; Lin, R.; Mossman, K.L.; Zheng, C. Herpes simplex virus 1 tegument protein us11 downmodulates the rlr signaling pathway via direct interaction with rig-i and mda-5. J. Virol. 2012, 86, 3528–3540. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Jin, H.; Valyi-Nagy, T.; Cao, Y.; Yan, Z.; He, B. Inhibition of tank binding kinase 1 by herpes simplex virus 1 facilitates productive infection. J. Virol. 2012, 86, 2188–2196. [Google Scholar] [CrossRef] [PubMed]
- Verpooten, D.; Ma, Y.; Hou, S.; Yan, Z.; He, B. Control of tank-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J. Biol. Chem. 2009, 284, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Peri, P.; Mattila, R.K.; Kantola, H.; Broberg, E.; Karttunen, H.S.; Waris, M.; Vuorinen, T.; Hukkanen, V. Herpes simplex virus type 1 us3 gene deletion influences toll-like receptor responses in cultured monocytic cells. Virol. J. 2008, 5, 140. [Google Scholar] [CrossRef] [PubMed]
- Sen, J.; Liu, X.; Roller, R.; Knipe, D.M. Herpes simplex virus us3 tegument protein inhibits toll-like receptor 2 signaling at or before traf6 ubiquitination. Virology 2013, 439, 65–73. [Google Scholar] [CrossRef]
- Frame, M.C.; Purves, F.C.; McGeoch, D.J.; Marsden, H.S.; Leader, D.P. Identification of the herpes simplex virus protein kinase as the product of viral gene us3. J. Gen. Virol. 1987, 68, 2699–2704. [Google Scholar] [CrossRef]
- York, I.A.; Roop, C.; Andrews, D.W.; Riddell, S.R.; Graham, F.L.; Johnson, D.C. A cytosolic herpes simplex virus protein inhibits antigen presentation to cd8+ t lymphocytes. Cell 1994, 77, 525–535. [Google Scholar] [CrossRef]
- Oldham, M.L.; Hite, R.K.; Steffen, A.M.; Damko, E.; Li, Z.; Walz, T.; Chen, J. A mechanism of viral immune evasion revealed by cryo-em analysis of the tap transporter. Nature 2016, 529, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.H.; Jensen, S.B.; Miettinen, J.J.; Luecke, S.; Prabakaran, T.; Reinert, L.S.; Mettenleiter, T.; Chen, Z.J.; Knipe, D.M.; Sandri-Goldin, R.M.; et al. Hsv-1 icp27 targets the tbk1-activated sting signalsome to inhibit virus-induced type i ifn expression. EMBO J. 2016, 35, 1385–1399. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Micrornas: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Cullen, B.R. Micrornas as mediators of viral evasion of the immune system. Nat. Immunol. 2013, 14, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Baltimore, D. Micrornas as regulatory elements in immune system logic. Nat. Rev. Immunol. 2016, 16, 279–294. [Google Scholar] [CrossRef]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of virus-encoded micrornas. Science 2004, 304, 734–736. [Google Scholar] [CrossRef]
- Grey, F.; Antoniewicz, A.; Allen, E.; Saugstad, J.; McShea, A.; Carrington, J.C.; Nelson, J. Identification and characterization of human cytomegalovirus-encoded micrornas. J. Virol. 2005, 79, 12095–12099. [Google Scholar] [CrossRef]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grässer, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identification of micrornas of the herpesvirus family. Nature Methods 2005, 2, 269–276. [Google Scholar] [CrossRef]
- Bai, Z.-q.; Lei, X.-f.; Wang, L.-d.; Gao, S.-j. Identification and function of micrornas encoded by herpesviruses. Virol. Sin. 2008, 23, 459–472. [Google Scholar] [CrossRef]
- Grey, F.; Hook, L.; Nelson, J. The functions of herpesvirus-encoded micrornas. Med. Microbiol. Immunol. 2008, 197, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Griffiths, A.; Li, G.; Silva, L.M.; Kramer, M.F.; Gaasterland, T.; Wang, X.J.; Coen, D.M. Prediction and identification of herpes simplex virus 1-encoded micrornas. J. Virol. 2006, 80, 5499–5508. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. Micrornas expressed by herpes simplex virus 1 during latent infection regulate viral mrnas. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Boss, I.W.; Plaisance, K.B.; Renne, R. Role of virus-encoded micrornas in herpesvirus biology. Trends Microbiol. 2009, 17, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. Mirbase: Annotating high confidence micrornas using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Han, Z.; Zhou, G.; Roizman, B. Patterns of accumulation of mirnas encoded by herpes simplex virus during productive infection, latency, and on reactivation. Proc. Natl Acad. Sci. USA 2015, 112, E49–E55. [Google Scholar] [CrossRef] [PubMed]
- Flores, O.; Nakayama, S.; Whisnant, A.W.; Javanbakht, H.; Cullen, B.R.; Bloom, D.C. Mutational inactivation of herpes simplex virus 1 micrornas identifies viral mrna targets and reveals phenotypic effects in culture. J. Virol. 2013, 87, 6589–6603. [Google Scholar] [CrossRef] [PubMed]
- Enk, J.; Levi, A.; Weisblum, Y.; Yamin, R.; Charpak-Amikam, Y.; Wolf, D.G.; Mandelboim, O. Hsv1 microrna modulation of gpi anchoring and downstream immune evasion. Cell Rep. 2016, 17, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Guo, Z.; Zhang, X.; Guo, L.; Liu, L.; Liao, Y.; Wang, J.; Wang, L.; Li, Q. A microrna encoded by hsv-1 inhibits a cellular transcriptional repressor of viral immediate early and early genes. Sci. China Life Sci. 2013, 56, 373–383. [Google Scholar] [CrossRef]
- Han, Z.; Liu, X.; Chen, X.; Zhou, X.; Du, T.; Roizman, B.; Zhou, G. Mir-h28 and mir-h29 expressed late in productive infection are exported and restrict hsv-1 replication and spread in recipient cells. Proc. Natl Acad. Sci. USA 2016, 113, E894–E901. [Google Scholar] [CrossRef]
- Mestdagh, P.; Feys, T.; Bernard, N.; Guenther, S.; Chen, C.; Speleman, F.; Vandesompele, J. High-throughput stem-loop rt-qpcr mirna expression profiling using minute amounts of input rna. Nucleic Acids Res. 2008, 36, e143. [Google Scholar] [CrossRef] [PubMed]
- Rehmsmeier, M.; Steffen, P.; Hochsmann, M.; Giegerich, R. Fast and effective prediction of microrna/target duplexes. Rna 2004, 10, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Kruger, J.; Rehmsmeier, M. Rnahybrid: Microrna target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Sharp, P.A. Specificity of microrna target selection in translational repression. Genes Dev. 2004, 18, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Bao, M.; Lu, N.; Weng, L.; Yuan, B.; Liu, Y.J. The e3 ubiquitin ligase trim21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nat. Immunol. 2013, 14, 172–178. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Conwell, S.E.; Berrios, C.; DeCaprio, J.A.; Knipe, D.M. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc. Natl Acad. Sci. USA 2013, 110, E4492–E4501. [Google Scholar] [CrossRef]
- Pan, D.; Flores, O.; Umbach, J.L.; Pesola, J.M.; Bentley, P.; Rosato, P.C.; Leib, D.A.; Cullen, B.R.; Coen, D.M. A neuron-specific host microrna targets herpes simplex virus-1 icp0 expression and promotes latency. Cell Host Microbe. 2014, 15, 446–456. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Broekema, N.M.; Knipe, D.M. Relative contributions of herpes simplex virus 1 icp0 and vhs to loss of cellular ifi16 vary in different human cell types. J. Virol. 2016, 90, 8351–8359. [Google Scholar] [CrossRef]
- Pan, S.; Liu, X.; Ma, Y.; Cao, Y.; He, B. Herpes simplex virus 1 γ1 34.5 protein inhibits sting activation that restricts viral replication. J. Virol. 2018, 92, e01015-18. [Google Scholar] [CrossRef]
- Deschamps, T.; Kalamvoki, M. Evasion of the sting DNA-sensing pathway by vp11/12 of herpes simplex virus 1. J. Virol. 2017, 91, e00535-17. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, J.; Xu, S.; Li, J.; He, S.; Zeng, Y.; Xie, L.; Xie, N.; Liu, T.; Lee, K.; et al. Species-specific deamidation of cgas by herpes simplex virus ul37 protein facilitates viral replication. Cell Host Microbe. 2018, 24, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Zheng, C. Herpes simplex virus 1 abrogates the cgas/sting-mediated cytosolic DNA-sensing pathway via its virion host shutoff protein, ul41. J. Virol. 2017, 91, e02414-16. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; You, H.; Su, C.; Li, Y.; Chen, S.; Zheng, C. Herpes simplex virus 1 tegument protein vp22 abrogates cgas/sting-mediated antiviral innate immunity. J. Virol. 2018, 92, e00841-18. [Google Scholar] [CrossRef] [PubMed]
- Maruzuru, Y.; Ichinohe, T.; Sato, R.; Miyake, K.; Okano, T.; Suzuki, T.; Koshiba, T.; Koyanagi, N.; Tsuda, S.; Watanabe, M.; et al. Herpes simplex virus 1 vp22 inhibits aim2-dependent inflammasome activation to enable efficient viral replication. Cell Host Microbe. 2018, 23, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Cokaric Brdovcak, M.; Zubkovic, A.; Jurak, I. Herpes simplex virus 1 deregulation of host micrornas. Non-coding RNA 2018, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Hu, N.; Mo, L.; Zeng, Z.; Sun, J.; Hu, Y. Deep rna sequencing reveals a repertoire of human fibroblast circular rnas associated with cellular responses to herpes simplex virus 1 infection. Cell Physiol. Biochem. 2018, 47, 2031–2045. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, B.; Esclatine, A.; Zhang, W.; Roizman, B. The stress-inducible immediate-early responsive gene iex-1 is activated in cells infected with herpes simplex virus 1, but several viral mechanisms, including 3′ degradation of its rna, preclude expression of the gene. J. Virol. 2003, 77, 6178–6187. [Google Scholar] [CrossRef] [PubMed]
- Shu, M.; Taddeo, B.; Roizman, B. Tristetraprolin recruits the herpes simplex virion host shutoff rnase to au-rich elements in stress response mrnas to enable their cleavage. J. Virol. 2015, 89, 5643–5650. [Google Scholar] [CrossRef]
- Mignone, F.; Pesole, G. Mrna Untranslated Regions (utrs); John Wiley & Sons: Chichester, England, 2011; pp. 1–5. [Google Scholar]
- Asirvatham, A.J.; Gregorie, C.J.; Hu, Z.; Magner, W.J.; Tomasi, T.B. Microrna targets in immune genes and the dicer/argonaute and are machinery components. Mol. Immunol. 2008, 45, 1995–2006. [Google Scholar] [CrossRef]
- Zhang, R.; Su, B. Small but influential: The role of micrornas on gene regulatory network and 3′utr evolution. J. Genet. Genom. 2009, 36, 1–6. [Google Scholar] [CrossRef]
- Xu, L.; Peng, L.; Gu, T.; Yu, D.; Yao, Y.G. The 3′utr of human mavs mrna contains multiple regulatory elements for the control of protein expression and subcellular localization. Biochim. Biophys. Acta. Gene Regul. Mech. 2019, 1862, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Brown, D.; Osorio, N.; Hsiang, C.; BenMohamed, L.; Wechsler, S.L. Increased neurovirulence and reactivation of the herpes simplex virus type 1 latency-associated transcript (lat)-negative mutant dlat2903 with a disrupted lat mir-h2. J. Neurovirol. 2016, 22, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Brown, D.; Osorio, N.; Hsiang, C.; Li, L.; Chan, L.; BenMohamed, L.; Wechsler, S.L. A herpes simplex virus type 1 mutant disrupted for microrna h2 with increased neurovirulence and rate of reactivation. J. Neurovirol. 2015, 21, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Pesola, J.M.; Li, G.; McCarron, S.; Coen, D.M.; Sandri-Goldin, R.M. Mutations inactivating herpes simplex virus 1 microrna mir-h2 do not detectably increase icp0 gene expression in infected cultured cells or mouse trigeminal ganglia. J. Virol. 2017, 91, e02001–e02016. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.G.; Kim, S.S.; Kui, L.; Voon, D.C.; Mauduit, M.; Bist, P.; Bi, X.; Pereira, N.A.; Liu, C.; Sukumaran, B.; et al. Bruton’s tyrosine kinase phosphorylates ddx41 and activates its binding of dsdna and sting to initiate type 1 interferon response. Cell Rep. 2015, 10, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Parvatiyar, K.; Zhang, Z.; Teles, R.M.; Ouyang, S.; Jiang, Y.; Iyer, S.S.; Zaver, S.A.; Schenk, M.; Zeng, S.; Zhong, W.; et al. The helicase ddx41 recognizes the bacterial secondary messengers cyclic di-gmp and cyclic di-amp to activate a type i interferon immune response. Nat. Immunol. 2012, 13, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Astor, T.L.; Liptak, L.M.; Cho, C.; Coen, D.M.; Schaffer, P.A. The herpes simplex virus type 1 regulatory protein icp0 enhances virus replication during acute infection and reactivation from latency. J. Virol. 1993, 67, 7501–7512. [Google Scholar] [PubMed]
- Everett, R.D. Icp0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays 2000, 22, 761–770. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primers (5′→3′) |
|---|---|
| DDX41 | Forward: GAGCTCGCCGACAGTCTTCCCTTCTC |
| Reverse: TCTAGATGGGCTAGAGGTTTGGGCTT | |
| IFI16 | Forward: GAGCTCAATCTGGATGTCATTGACGA |
| Reverse: TCTAGAATGTTTATTTATTTTTTCAA | |
| DAI | Forward: GAGCTCGCCGACAGTCTTCCCTTCTC |
| Reverse: TCTAGAATGGTTGGTTTATTTTTTAT | |
| cGAS | Forward: GAGCTCGATTGTATTTTTAGAAAGAT |
| Reverse: TCTAGAGTGAGCCACAGCGTCT | |
| STING | Forward: GAGCTCGACCCAGGGTCACCAGGCCA |
| Reverse: TCTAGACCGTTGAGAAAGGGGTGGAG | |
| TRIF | Forward: GAGCTCCCGCGTGTCCTTGCCTGACC |
| Reverse: TCTAGAGCTGCGGAAGCATTCAATAA | |
| TBK1 | Forward: GAGCTCTTTCTAATAGAAGTTTAAGA |
| Reverse: TCTAGATTCCATGAAATAGTAAGCAG | |
| TLR9 | Forward: GAGCTCCCGTGAGCCGGAATCCTGCA |
| Reverse: TCTAGATGCTCTGTGTCAGGTGTGGG | |
| IRF3 | Forward: GAGCTCGCCCTCGCTCCTCATGGTGT |
| Reverse: TCTAGATTTGATCATAGCAGGAACCA | |
| IRF7 | Forward: GAGCTCAACCCAGTCTAATGAGAACT |
| Reverse: TCTAGATGTTCTGGAGTTCTTTTTAT | |
| IRF9 | Forward: GAGCTCAGCCTGGGGGACCCATCTTC |
| Reverse: TCTAGAAAGTGCTTGGCTTTAGAGTT | |
| UL30 | Forward: CATCACCGACCCGGAGAGGGAC |
| Reverse: GGGCCAGGCGCTTGTTGGTGTA | |
| hsa-miR-16 | Sense: CGCCAATATTTACGTGCTGCTA |
| Antisense: CTAGTAGCAGCACGTAAATATTGGCGAGCT |
| Gene | Sequences (5′→3′) |
|---|---|
| DDX41 | Sense: GAUGGCUAAGGGCAUUACGTT |
| Antisense: CGUAAUGCCCUUAGCCAUCTT | |
| IFI16 | Sense: TAGCGTTTCTGGAGATTACAATT |
| Antisense: UUGUAAUCUCCAGAAACGCUATT | |
| Scramble Control | Sense: CUAAUCAGGCGAUGAAUGUTT |
| Antisense: ACAUUCAUCGCCUGAUUAGTT |
| Gene | Sequences (5′→3′) |
|---|---|
| miR-H2-3p mimics | Sense: CCUGAGCCAGGGACGAGUGCGACU |
| Antisense: UCGCACUCGUCCCUGGCUCAGGUU | |
| miR-H2-3p mimics NC | Sense: UUCUCCGAACGUGUCACGUTT |
| Antisense: ACGUGACACGUUCGGAGAATT | |
| miR-H2-3p inhibitors | AGUCGCACUCGUCCCUGGCUCAGG |
| miR-H2-3p inhibitors NC | CAGUACUUUUGUGUAGUACAA |
| Gene | Primers (5′→3′) |
|---|---|
| DDX41 | Forward: GACACTGGTGTTCACGTTGC |
| Reverse: CGAGGGGCAGATGATGAGTC | |
| IFI16 | Forward: AGAAACAATGACCCCAAGAGC |
| Reverse: CTTGGTGAAGAAACTGCTGGAT | |
| IFN-β | Forward: CCAACAAGTGTCTCCTCCAA |
| Reverse: ATAGTCTCATTCCAGCCAGT | |
| MxI | Forward: CTGAAGGAGCGGAGCGACAC |
| Reverse: TTAACCGGGGAACTGGGCAA | |
| GAPDH | Forward: CATGTTCGTCATGGGTGTGAACCA |
| Reverse: AGTGATGGCATGGACTGTGGTCAT | |
| TK | Forward: GATGGGGAAAACCACCACCA |
| Reverse: TGTTCGCGATTGTCTCGGAA | |
| gC | Forward: CCAACCTAACGGACCCCCAC |
| Reverse: GCGTCACCTCGCCGATAATC | |
| ICP0 | Forward: ACACGGCAGCAGTAACACCA |
| Reverse: TCCGCTTCAACAACCCCAAC | |
| miR-H2-3p | RT: GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAGTCGCA |
| Forward: CCCTGAGCCAGGGACGAG | |
| Reverse: CCAGTGCAGGGTCCGAGGTA | |
| U6 snoRNA | Forward: GCTCGCTTCGGCAGCACATA |
| Reverse: TGGAACGCTTCACGAATTTG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, Y.; Zeng, J.; Fan, S.; Liao, Y.; Feng, M.; Wang, L.; Zhang, Y.; Li, Q. Herpes Simplex Virus Type 1–Encoded miR-H2-3p Manipulates Cytosolic DNA–Stimulated Antiviral Innate Immune Response by Targeting DDX41. Viruses 2019, 11, 756. https://doi.org/10.3390/v11080756
Duan Y, Zeng J, Fan S, Liao Y, Feng M, Wang L, Zhang Y, Li Q. Herpes Simplex Virus Type 1–Encoded miR-H2-3p Manipulates Cytosolic DNA–Stimulated Antiviral Innate Immune Response by Targeting DDX41. Viruses. 2019; 11(8):756. https://doi.org/10.3390/v11080756
Chicago/Turabian StyleDuan, Yongzhong, Jieyuan Zeng, Shengtao Fan, Yun Liao, Min Feng, Lichun Wang, Ying Zhang, and Qihan Li. 2019. "Herpes Simplex Virus Type 1–Encoded miR-H2-3p Manipulates Cytosolic DNA–Stimulated Antiviral Innate Immune Response by Targeting DDX41" Viruses 11, no. 8: 756. https://doi.org/10.3390/v11080756
APA StyleDuan, Y., Zeng, J., Fan, S., Liao, Y., Feng, M., Wang, L., Zhang, Y., & Li, Q. (2019). Herpes Simplex Virus Type 1–Encoded miR-H2-3p Manipulates Cytosolic DNA–Stimulated Antiviral Innate Immune Response by Targeting DDX41. Viruses, 11(8), 756. https://doi.org/10.3390/v11080756