Aichi Virus Induces Antiviral Host Defense in Primary Murine Intestinal Epithelial Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus, Cell Lines, and Reagents
2.2. Isolation of Mouse Intestinal Epithelial Cells
2.3. Aichi Virus Infection and Plaque Assay
2.4. Immunofluorescence Assay
2.5. Quantitative Reverse Transcription-Polymerase Chain Reaction(qRT-PCR)
2.6. Western Blot Analysis
2.7. Cell Viability and Cytotoxicity Assay
2.8. Statistical Analysis
3. Results
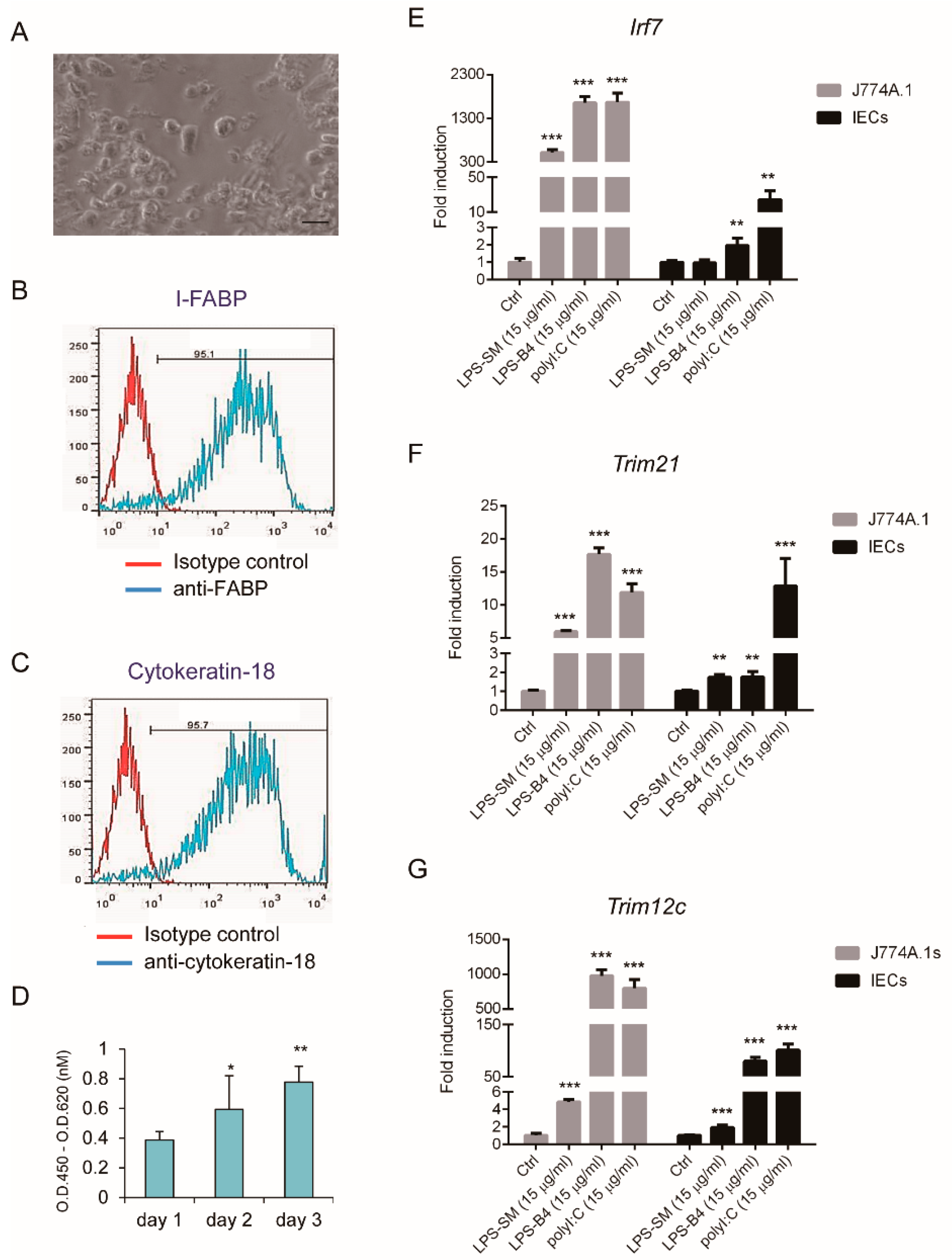
3.1. Isolation of Primary Mouse Intestinal Epithelial Cells for Toll-Like receptor-Ligand Stimulation
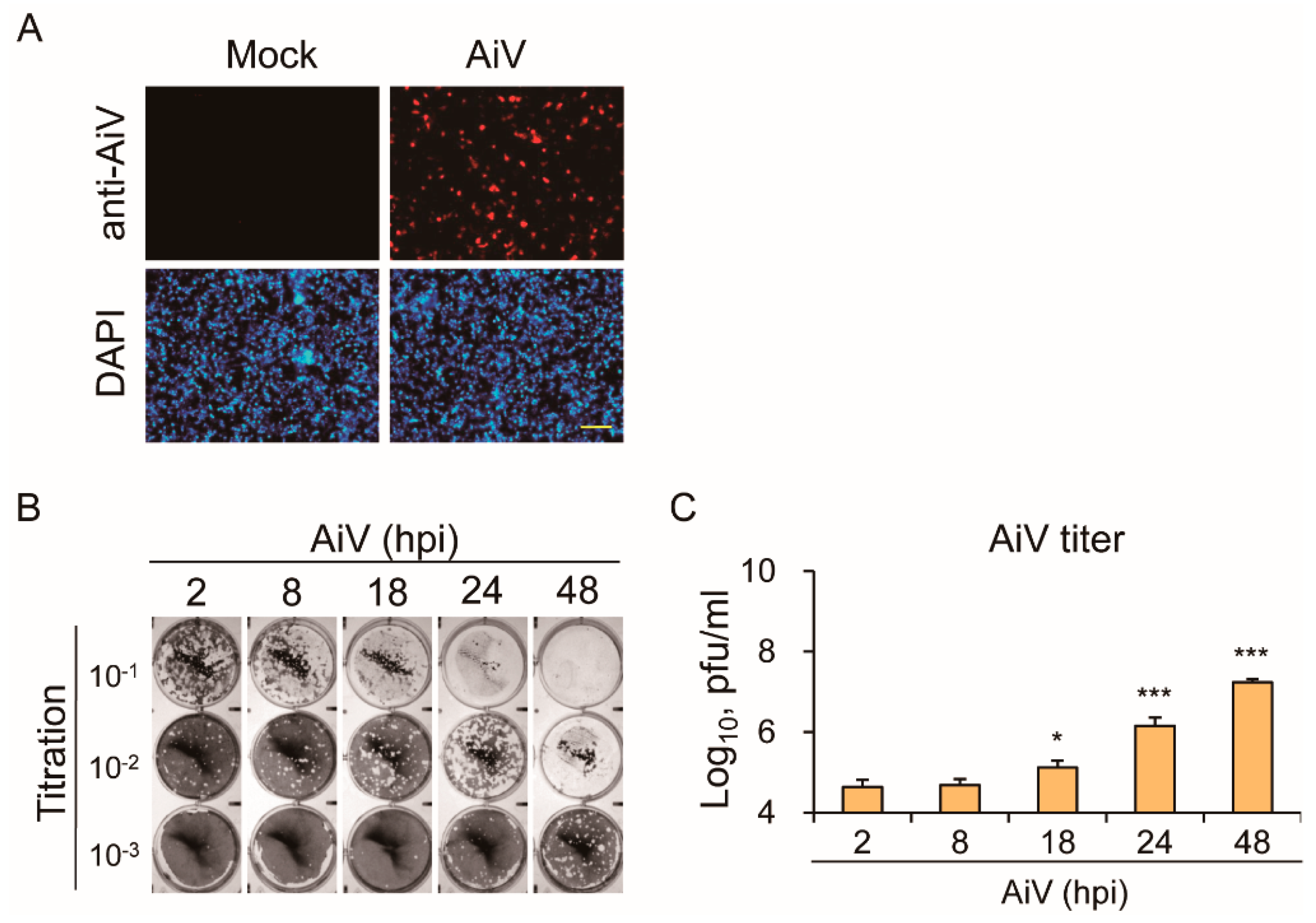
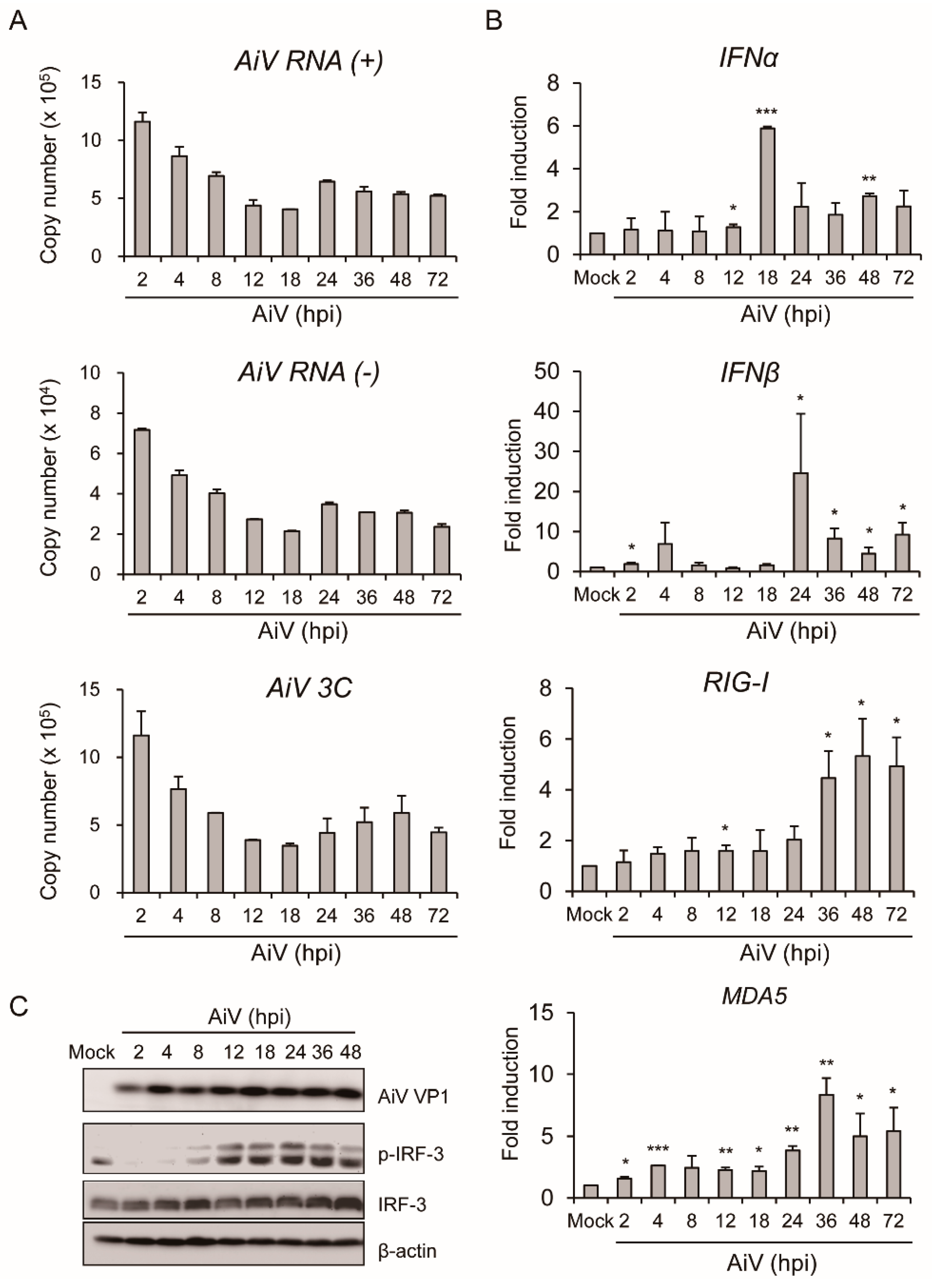
3.2. Productive Infection of Aichi Virus in Intestinal Epithelial Cells
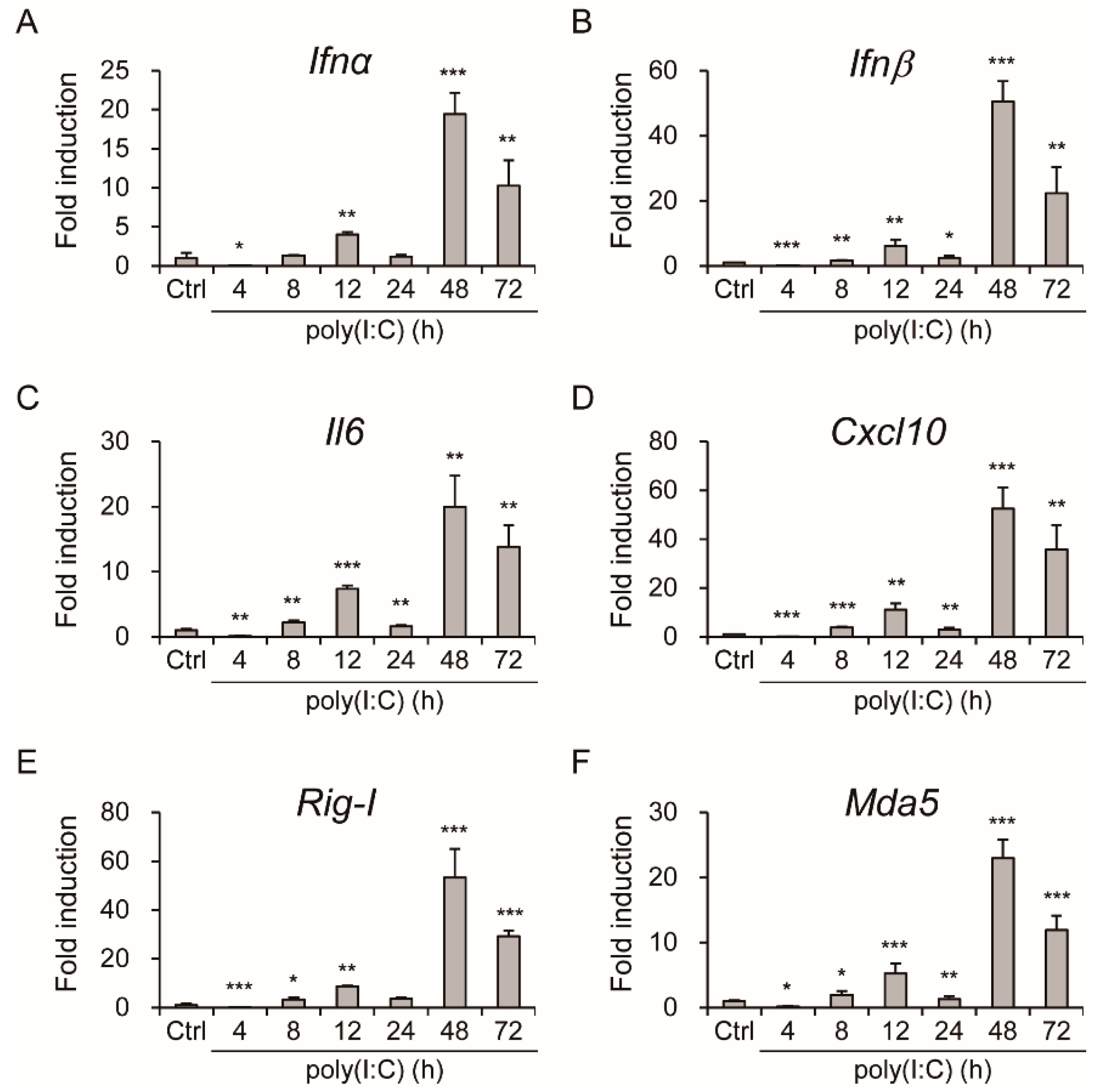
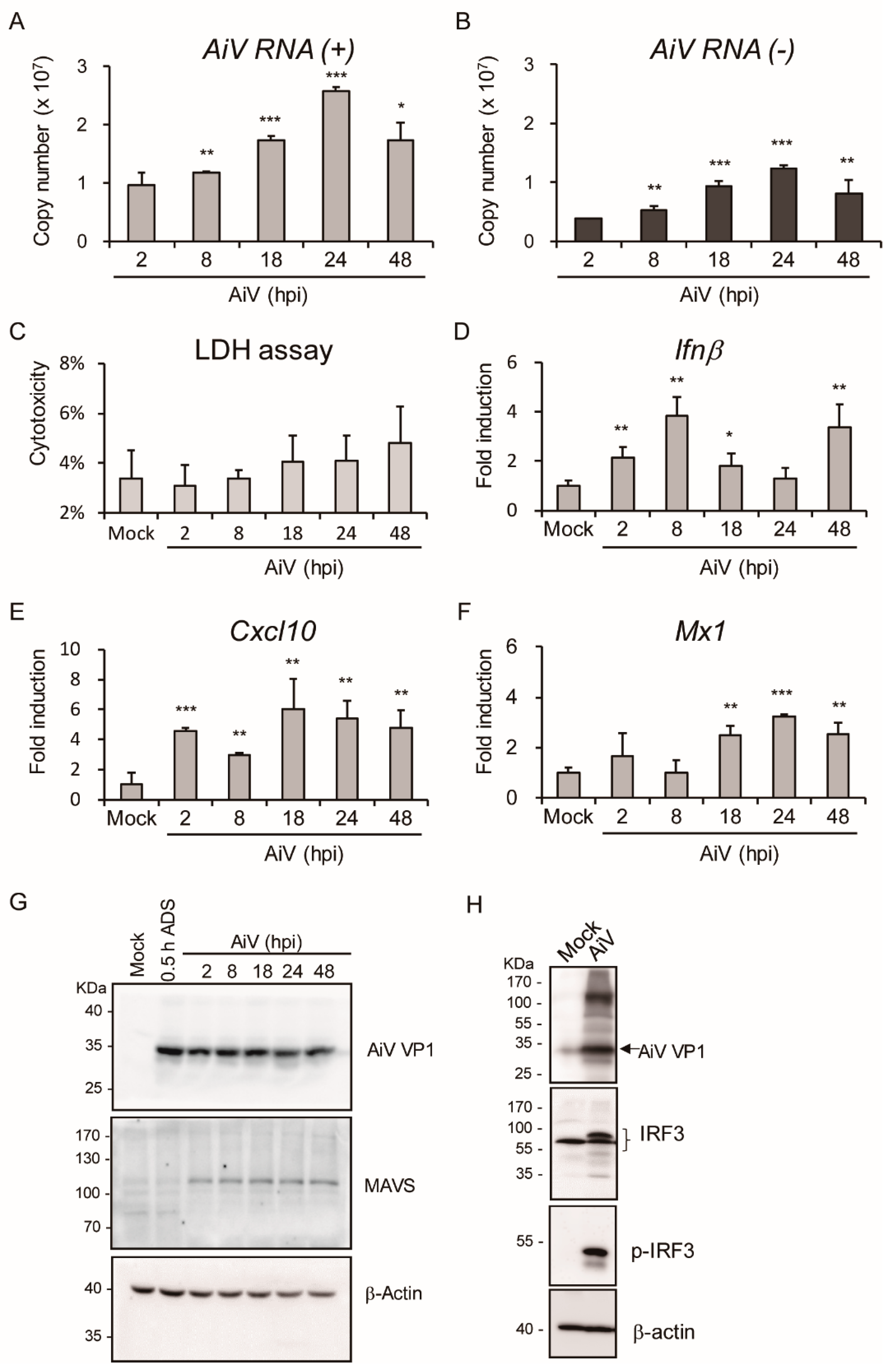
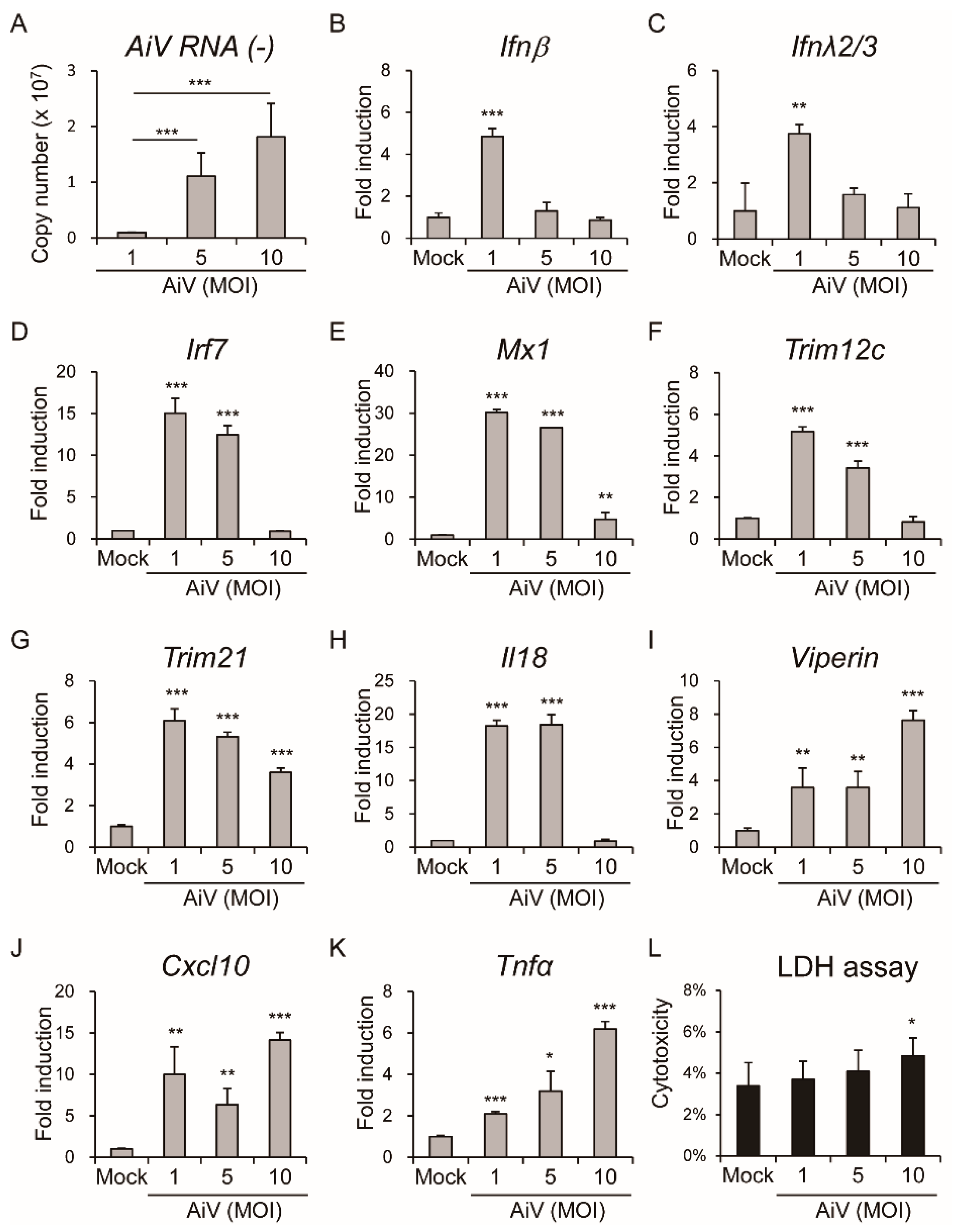
3.3. Aichi Virus-Induced Type I Interferons and Expression of Inflammatory Cytokines in Intestinal Epithelial Cells
3.4. Aichi Virus Activated Antiviral Response in T84 Human Intestinal Epithelial Cells
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Le Guyader, F.S.; Le Saux, J.C.; Ambert-Balay, K.; Krol, J.; Serais, O.; Parnaudeau, S.; Giraudon, H.; Delmas, G.; Pommepuy, M.; Pothier, P.; et al. Aichi virus, norovirus, astrovirus, enterovirus, and rotavirus involved in clinical cases from a French oyster-related gastroenteritis outbreak. J. Clin. Microbiol. 2008, 46, 4011–4017. [Google Scholar] [CrossRef]
- Yip, C.C.; Lo, K.L.; Que, T.L.; Lee, R.A.; Chan, K.H.; Yuen, K.Y.; Woo, P.C.; Lau, S.K. Epidemiology of human parechovirus, Aichi virus and salivirus in fecal samples from hospitalized children with gastroenteritis in Hong Kong. Virol. J. 2014, 11, 182. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Ito, M.; Tsuzuki, H.; Sakae, K. Identification of Aichi virus infection by measurement of immunoglobulin responses in an enzyme-linked immunosorbent assay. J. Clin. Microbiol. 2001, 39, 4178–4180. [Google Scholar] [CrossRef] [PubMed]
- Chuchaona, W.; Khamrin, P.; Yodmeeklin, A.; Kumthip, K.; Saikruang, W.; Thongprachum, A.; Okitsu, S.; Ushijima, H.; Maneekarn, N. Detection and characterization of Aichi virus 1 in pediatric patients with diarrhea in Thailand. J. Med. Virol. 2017, 89, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Reuter, G.; Boldizsar, A.; Papp, G.; Pankovics, P. Detection of Aichi virus shedding in a child with enteric and extraintestinal symptoms in Hungary. Arch. Virol. 2009, 154, 1529–1532. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Sakae, K.; Ishihara, Y.; Isomura, S.; Utagawa, E. Prevalence of newly isolated, cytopathic small round virus (Aichi strain) in Japan. J. Clin. Microbiol. 1993, 31, 2938–2943. [Google Scholar] [PubMed]
- Reuter, G.; Boros, A.; Pankovics, P. Kobuviruses—A comprehensive review. Rev. Med. Virol. 2011, 21, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Khamrin, P.; Maneekarn, N.; Okitsu, S.; Ushijima, H. Epidemiology of human and animal kobuviruses. Virusdisease 2014, 25, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Van Dung, N.; Ivens, A.; Bogaardt, C.; O’Toole, A.; Bryant, J.E.; Carrique-Mas, J.; Van Cuong, N.; Anh, P.H.; Rabaa, M.A.; et al. Genetic diversity and cross-species transmission of kobuviruses in Vietnam. Virus Evol. 2018, 4, vey002. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Kobayashi, S.; Sakae, K.; Nakata, S.; Chiba, S.; Ishihara, Y.; Isomura, S. Isolation of cytopathic small round viruses with BS-C-1 cells from patients with gastroenteritis. J. Infect. Dis. 1991, 164, 954–957. [Google Scholar] [CrossRef]
- Kitajima, M.; Gerba, C.P. Aichi virus 1: Environmental occurrence and behavior. Pathogens 2015, 4, 256–268. [Google Scholar] [CrossRef]
- Terio, V.; Bottaro, M.; Di Pinto, A.; Fusco, G.; Barresi, T.; Tantillo, G.; Martella, V. Occurrence of Aichi virus in retail shellfish in Italy. Food Microbiol. 2018, 74, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Chen, B.C.; Lin, Y.S.; Chang, J.T.; Huang, T.S.; Chen, J.J.; Chang, T.H. Detection of Aichi virus with antibody targeting of conserved viral protein 1 epitope. Appl. Microbiol. Biotechnol. 2013, 97, 8529–8536. [Google Scholar] [CrossRef]
- Chang, T.H.; Liao, C.L.; Lin, Y.L. Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-kappaB activation. Microbes Infect. 2006, 8, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Yang, C.S.; Chen, Y.S.; Chen, B.C.; Chiang, A.J.; Chang, Y.H.; Tsai, W.L.; Lin, Y.S.; Chao, D.; Chang, T.H. Genome and infection characteristics of human parechovirus type 1: The interplay between viral infection and type I interferon antiviral system. PLoS ONE 2015, 10, e0116158. [Google Scholar] [CrossRef]
- Schulz, K.S.; Mossman, K.L. Viral Evasion Strategies in Type I IFN Signaling—A Summary of Recent Developments. Front. Immunol. 2016, 7, 498. [Google Scholar] [CrossRef]
- Ingle, H.; Peterson, S.T.; Baldridge, M.T. Distinct Effects of Type I and III Interferons on Enteric Viruses. Viruses 2018, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Ning, S.; Pagano, J.S.; Barber, G.N. IRF7: Activation, regulation, modification and function. Genes Immun. 2011, 12, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C., 3rd. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Shi, H.X.; Liu, X.Y.; Shan, Y.F.; Wei, B.; Chen, S.; Wang, C. TRIM21 is essential to sustain IFN regulatory factor 3 activation during antiviral response. J. Immunol. 2009, 182, 3782–3792. [Google Scholar] [CrossRef]
- Chang, T.H.; Yoshimi, R.; Ozato, K. Tripartite Motif (TRIM) 12c, a Mouse Homolog of TRIM5, Is a Ubiquitin Ligase That Stimulates Type I IFN and NF-kappaB Pathways along with TNFR-Associated Factor 6. J. Immunol. 2015, 195, 5367–5379. [Google Scholar] [CrossRef] [PubMed]
- Broquet, A.H.; Hirata, Y.; McAllister, C.S.; Kagnoff, M.F. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J. Immunol. 2011, 186, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.L.; Chang, T.H.; Liao, C.L.; Lin, Y.L. The cellular antiviral protein viperin is attenuated by proteasome-mediated protein degradation in Japanese encephalitis virus-infected cells. J. Virol. 2008, 82, 10455–10464. [Google Scholar] [CrossRef]
- Haller, O.; Staeheli, P.; Kochs, G. Interferon-induced Mx proteins in antiviral host defense. Biochimie 2007, 89, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Trifilo, M.J.; Montalto-Morrison, C.; Stiles, L.N.; Hurst, K.R.; Hardison, J.L.; Manning, J.E.; Masters, P.S.; Lane, T.E. CXC chemokine ligand 10 controls viral infection in the central nervous system: Evidence for a role in innate immune response through recruitment and activation of natural killer cells. J. Virol. 2004, 78, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.H.; Chen, S.R.; Yu, C.Y.; Lin, Y.S.; Chen, Y.S.; Kubota, T.; Matsuoka, M.; Lin, Y.L. Dengue virus serotype 2 blocks extracellular signal-regulated kinase and nuclear factor-kappaB activation to downregulate cytokine production. PLoS ONE 2012, 7, e41635. [Google Scholar]
- Yu, C.Y.; Chang, T.H.; Liang, J.J.; Chiang, R.L.; Lee, Y.L.; Liao, C.L.; Lin, Y.L. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog. 2012, 8, e1002780. [Google Scholar] [CrossRef]
- Chang, T.H.; Kubota, T.; Matsuoka, M.; Jones, S.; Bradfute, S.B.; Bray, M.; Ozato, K. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog. 2009, 5, e1000493. [Google Scholar] [CrossRef]
- Gribar, S.C.; Anand, R.J.; Sodhi, C.P.; Hackam, D.J. The role of epithelial Toll-like receptor signaling in the pathogenesis of intestinal inflammation. J. Leukoc. Biol. 2008, 83, 493–498. [Google Scholar] [CrossRef]
- Cetin, S.; Ford, H.R.; Sysko, L.R.; Agarwal, C.; Wang, J.; Neal, M.D.; Baty, C.; Apodaca, G.; Hackam, D.J. Endotoxin inhibits intestinal epithelial restitution through activation of Rho-GTPase and increased focal adhesions. J. Biol. Chem. 2004, 279, 24592–24600. [Google Scholar] [CrossRef]
- Leaphart, C.L.; Cavallo, J.; Gribar, S.C.; Cetin, S.; Li, J.; Branca, M.F.; Dubowski, T.D.; Sodhi, C.P.; Hackam, D.J. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J. Immunol. 2007, 179, 4808–4820. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.J.; Leaphart, C.L.; Mollen, K.P.; Hackam, D.J. The role of the intestinal barrier in the pathogenesis of necrotizing enterocolitis. Shock 2007, 27, 124–133. [Google Scholar] [CrossRef] [PubMed]
- McAllister, C.S.; Lakhdari, O.; Pineton de Chambrun, G.; Gareau, M.G.; Broquet, A.; Lee, G.H.; Shenouda, S.; Eckmann, L.; Kagnoff, M.F. TLR3, TRIF, and caspase 8 determine double-stranded RNA-induced epithelial cell death and survival in vivo. J. Immunol. 2013, 190, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Sun, Q.; Wang, S.; Zhang, Z.; Li, X.; Cardona, C.J.; Jin, Y.; Xing, Z. Robust antiviral responses to enterovirus 71 infection in human intestinal epithelial cells. Virus Res. 2013, 176, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Pott, J.; Mahlakoiv, T.; Mordstein, M.; Duerr, C.U.; Michiels, T.; Stockinger, S.; Staeheli, P.; Hornef, M.W. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 2011, 108, 7944–7949. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Zhu, S.; Ren, L.; Feng, N.; Song, Y.; Ge, X.; Li, B.; Flavell, R.A.; Greenberg, H.B. Rotavirus VP3 targets MAVS for degradation to inhibit type III interferon expression in intestinal epithelial cells. eLife 2018, 7, e3949. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Chen, Y.S.; Chen, B.C.; Chao, D.; Chang, T.H. Complete genome sequence of the first aichi virus isolated in taiwan. Genome Announc. 2013, 1, e00107-12. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Reikine, S.; Nguyen, J.B.; Modis, Y. Pattern Recognition and Signaling Mechanisms of RIG-I and MDA5. Front. Immunol. 2014, 5, 342. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef]
- Vijay-Kumar, M.; Gentsch, J.R.; Kaiser, W.J.; Borregaard, N.; Offermann, M.K.; Neish, A.S.; Gewirtz, A.T. Protein kinase R mediates intestinal epithelial gene remodeling in response to double-stranded RNA and live rotavirus. J. Immunol. 2005, 174, 6322–6331. [Google Scholar] [CrossRef]
- Metzger, R.N.; Krug, A.B.; Eisenacher, K. Enteric Virome Sensing-Its Role in Intestinal Homeostasis and Immunity. Viruses 2018, 10, 146. [Google Scholar] [CrossRef]
- Thuringer, D.; Berthenet, K.; Cronier, L.; Solary, E.; Garrido, C. Primary tumor- and metastasis-derived colon cancer cells differently modulate connexin expression and function in human capillary endothelial cells. Oncotarget 2015, 6, 28800–28815. [Google Scholar] [CrossRef] [Green Version]
- Failli, A.; Consolini, R.; Legitimo, A.; Spisni, R.; Castagna, M.; Romanini, A.; Crimaldi, G.; Miccoli, P. The challenge of culturing human colorectal tumor cells: Establishment of a cell culture model by the comparison of different methodological approaches. Tumori 2009, 95, 343–347. [Google Scholar] [CrossRef]
- Fernando, E.H.; Dicay, M.; Stahl, M.; Gordon, M.H.; Vegso, A.; Baggio, C.; Alston, L.; Lopes, F.; Baker, K.; Hirota, S.; et al. A simple, cost-effective method for generating murine colonic 3D enteroids and 2D monolayers for studies of primary epithelial cell function. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G467–G475. [Google Scholar] [CrossRef]
- Co, J.Y.; Margalef-Catala, M.; Li, X.; Mah, A.T.; Kuo, C.J.; Monack, D.M.; Amieva, M.R. Controlling Epithelial Polarity: A Human Enteroid Model for Host-Pathogen Interactions. Cell Rep. 2019, 26, 2509–2520.e4. [Google Scholar] [CrossRef] [Green Version]
- Drummond, C.G.; Bolock, A.M.; Ma, C.; Luke, C.J.; Good, M.; Coyne, C.B. Enteroviruses infect human enteroids and induce antiviral signaling in a cell lineage-specific manner. Proc. Natl. Acad. Sci. USA 2017, 114, 1672–1677. [Google Scholar] [CrossRef] [Green Version]
- Macartney, K.K.; Baumgart, D.C.; Carding, S.R.; Brubaker, J.O.; Offit, P.A. Primary murine small intestinal epithelial cells, maintained in long-term culture, are susceptible to rotavirus infection. J. Virol. 2000, 74, 5597–5603. [Google Scholar] [CrossRef]
- Yi, L.; He, Y.; Chen, Y.; Kung, H.F.; He, M.L. Potent inhibition of human enterovirus 71 replication by type I interferon subtypes. Antivir. Ther. 2011, 16, 51–58. [Google Scholar] [CrossRef]
- Sartor, R.B.; Wu, G.D. Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology 2017, 152, 327–339.e4. [Google Scholar] [CrossRef] [Green Version]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef]
- Baldridge, M.T.; Lee, S.; Brown, J.J.; McAllister, N.; Urbanek, K.; Dermody, T.S.; Nice, T.J.; Virgin, H.W. Expression of Ifnlr1 on Intestinal Epithelial Cells Is Critical to the Antiviral Effects of Interferon Lambda against Norovirus and Reovirus. J. Virol. 2017, 91, e02079-16. [Google Scholar] [CrossRef]
- Nice, T.J.; Baldridge, M.T.; McCune, B.T.; Norman, J.M.; Lazear, H.M.; Artyomov, M.; Diamond, M.S.; Virgin, H.W. Interferon-lambda cures persistent murine norovirus infection in the absence of adaptive immunity. Science 2015, 347, 269–273. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, Y.-T.; Kung, M.-H.; Hsu, T.-H.; Hung, W.-T.; Chen, Y.-S.; Yen, L.-C.; Chang, T.-H. Aichi Virus Induces Antiviral Host Defense in Primary Murine Intestinal Epithelial Cells. Viruses 2019, 11, 763. https://doi.org/10.3390/v11080763
Chang Y-T, Kung M-H, Hsu T-H, Hung W-T, Chen Y-S, Yen L-C, Chang T-H. Aichi Virus Induces Antiviral Host Defense in Primary Murine Intestinal Epithelial Cells. Viruses. 2019; 11(8):763. https://doi.org/10.3390/v11080763
Chicago/Turabian StyleChang, Yun-Te, Ming-Hsiang Kung, Thung-Hsien Hsu, Wan-Ting Hung, Yao-Shen Chen, Li-Chen Yen, and Tsung-Hsien Chang. 2019. "Aichi Virus Induces Antiviral Host Defense in Primary Murine Intestinal Epithelial Cells" Viruses 11, no. 8: 763. https://doi.org/10.3390/v11080763
APA StyleChang, Y. -T., Kung, M. -H., Hsu, T. -H., Hung, W. -T., Chen, Y. -S., Yen, L. -C., & Chang, T. -H. (2019). Aichi Virus Induces Antiviral Host Defense in Primary Murine Intestinal Epithelial Cells. Viruses, 11(8), 763. https://doi.org/10.3390/v11080763