Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
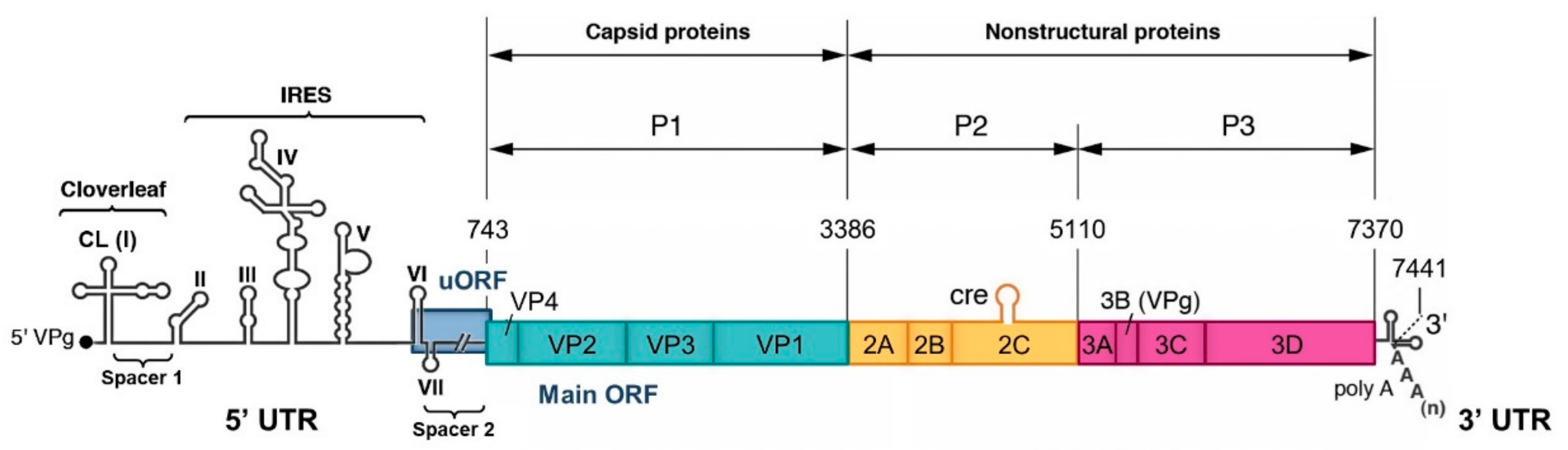
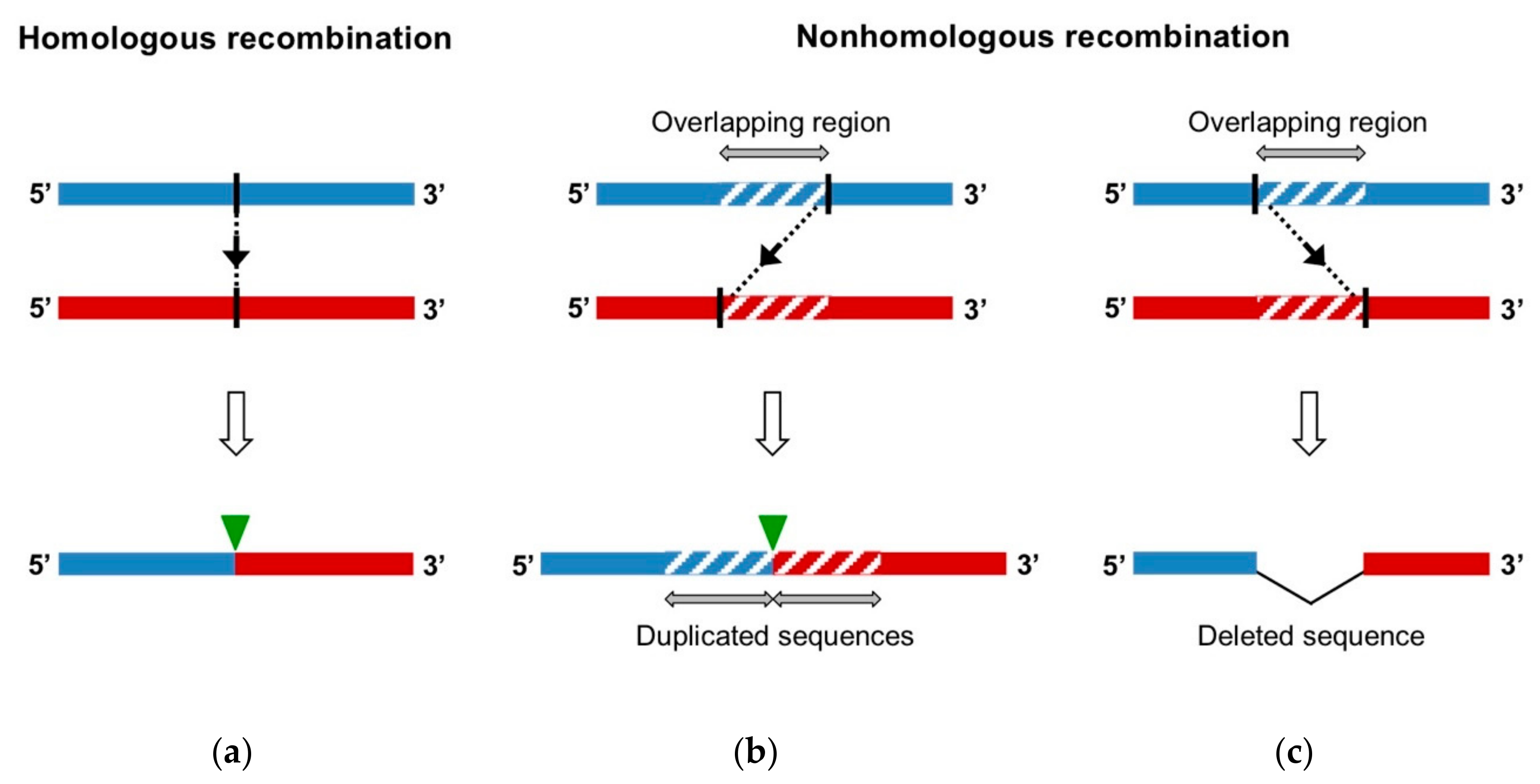
- Homologous recombination occurs at the same site in both parental genomes, therefore no insertion or deletion is observed at the recombination site when the recombinant genome is aligned with parental genomes.
- Nonhomologous recombination occurs at different sites in the two involved genetic fragments, generating aberrant structures, such as deletions or duplications of homologous parental sequences on each side of the recombination site.
2. Theoretical Model of Enterovirus Evolution Through Modular Intertypic Recombination
2.1. High Recombination Frequency in Enteroviruses
- Co-circulation: Several studies evaluating the circulation and genetic diversity of EVs in restricted geographic areas and on a short period of time revealed an extensive co-circulation of a high number of types from the four human EV species, usually associated with a high intra- and intertypic recombination frequency [65,92,93,94,95,96,97].Host co-infection: Consistent with the intense co-circulation observed, many cases of multiple infections in individuals have been reported [65,80,98].
- Cell co-infection: It was recently demonstrated that PV can spread as one unit containing multiple viral particles, either within lipid vesicles or as viral aggregates, and this delivery mode increased coinfection frequency and infectivity [99,100]. Furthermore, another recent study showed that certain resident bacteria of the gastrointestinal tract bind PV, increase viral co-infection of mammalian cells and enhance viral recombination, even when the ratio of virus to host cells is low, such as during the first cycle of replication following inter-host transmission [58].
- Colocalization of parental genomes: As all positive-strand RNA viruses, EVs replicate their genomes in virus-induced, membrane-bound replication compartments. The study of cells co-infected with two different PV strains showed that the majority of replication complexes contained both viral genomes, early in infection [101]. Moreover, a recent study in Brome mosaic virus (BMV) showed that the structure and size of the virus membranous replication compartments play a fundamental role on recruitment of multiple RNAs into a contiguous space, and thus on inter-genomic RNA recombination frequency, and accordingly suggested that the PV replication structures might favor RNA recombination [102,103].
- Selection: Finally, generated recombinant genomes have to be viable and able to efficiently compete with parental genomes and to confront bottleneck events occurring during virus life cycle, to spread in the viral population. This implies a structural and functional compatibility of the different recombining sequences, as well as a certain tolerance to genomic alterations in order to limit their negative consequences.
2.2. Analysis of Recombination Events in Circulating Enterovirus Strains
2.3. Genetic Features of Recombinant Circulating Vaccine-Derived Poliovirus Genomes
2.4. Modular Intertypic Recombination Hypothesis
3. Two Main Mechanisms of RNA Recombination in Enteroviruses
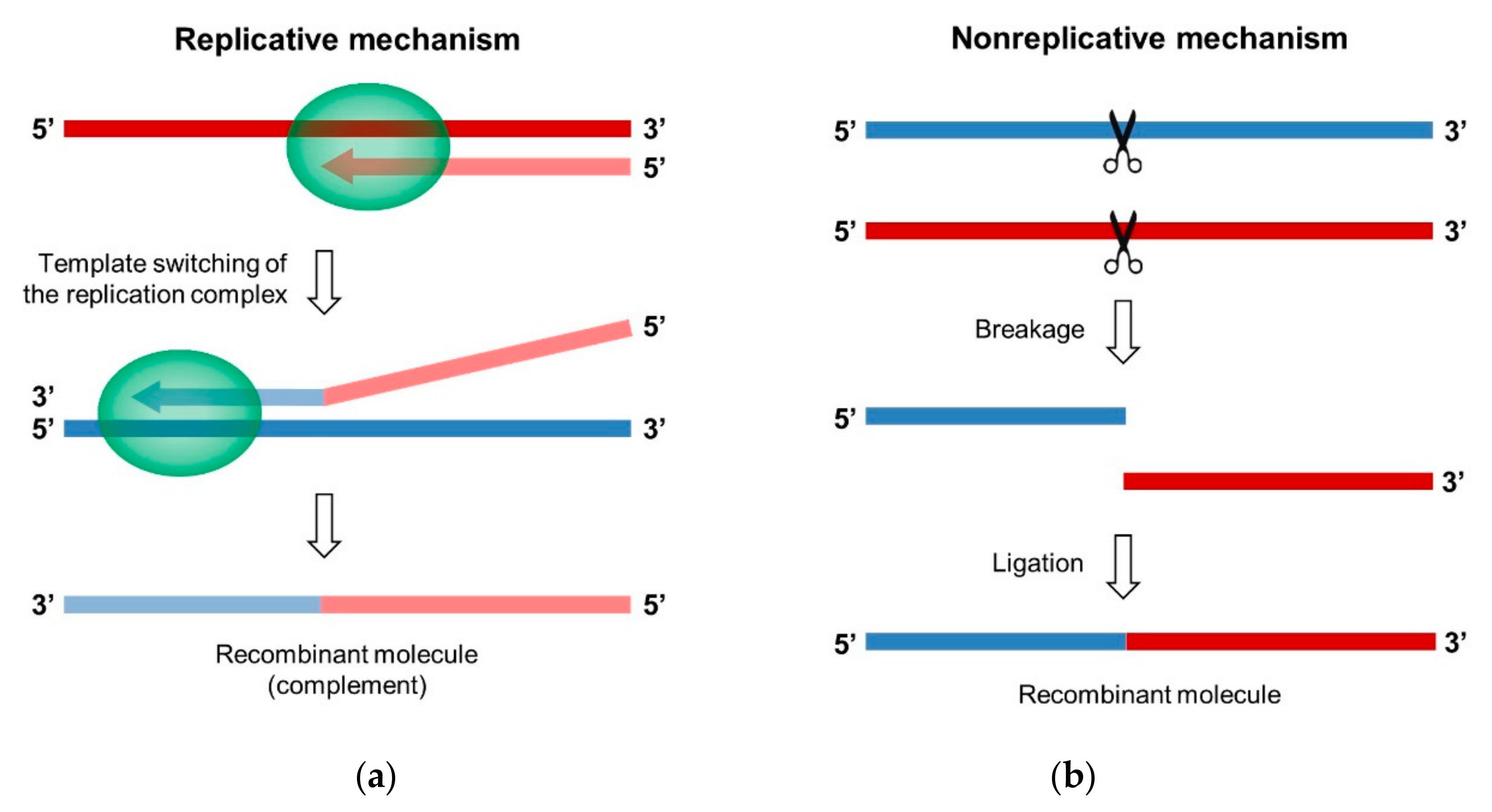
3.1. The Replicative Mechanism of Copy-Choice
3.1.1. Template Switching of the Viral Polymerase
3.1.2. Factors Influencing Template Switching
3.2. The Nonreplicative Mechanism of Breakage-Ligation
3.2.1. Demonstration in Poliovirus
3.2.2. Putative Implication of Cellular Factors
3.2.3. Alternative Mechanisms of RNA Recombination not Involving Viral RNA-Dependent RNA Polymerase
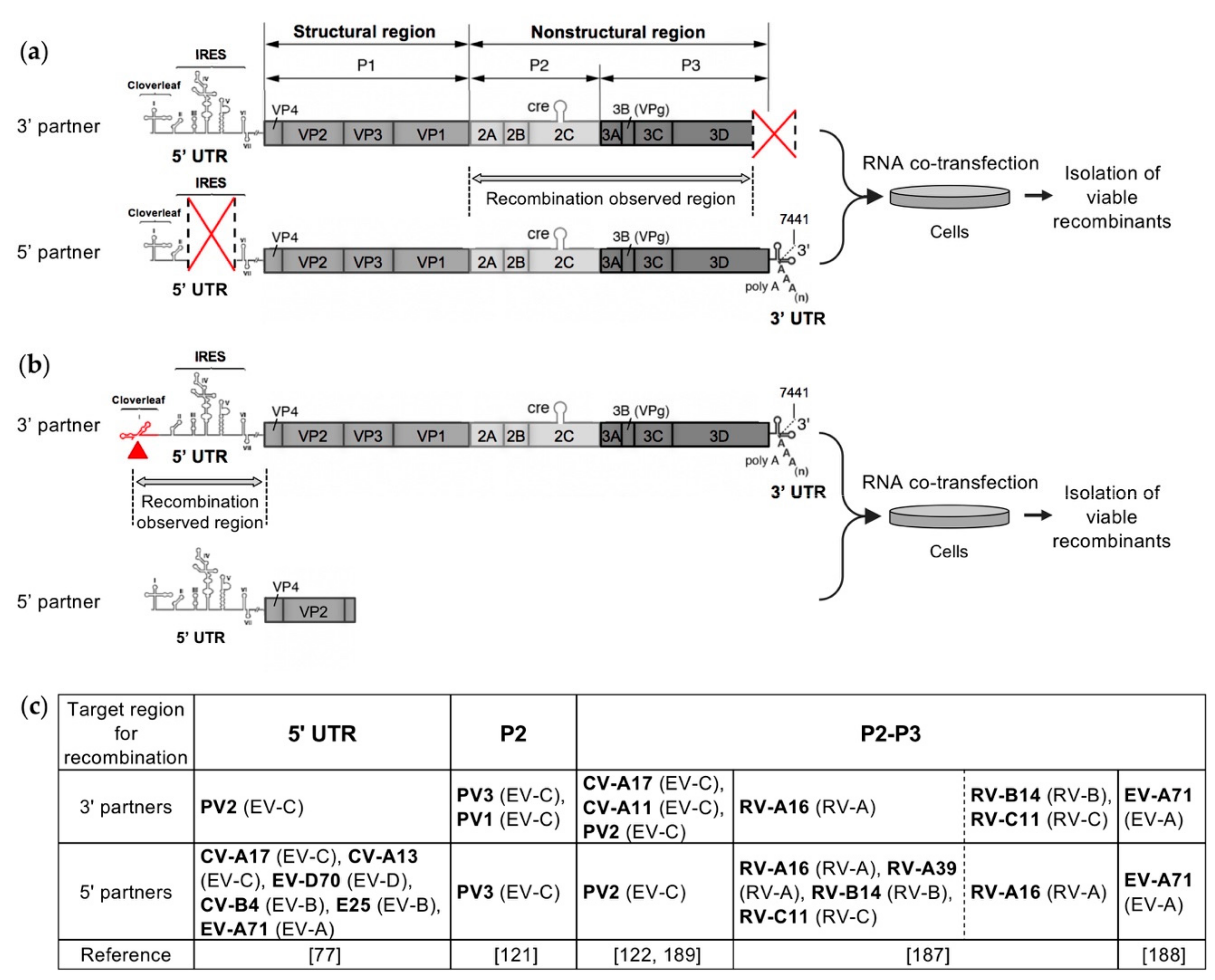
4. Recent Experimental Systems Designed to Study Recombination in Enteroviruses
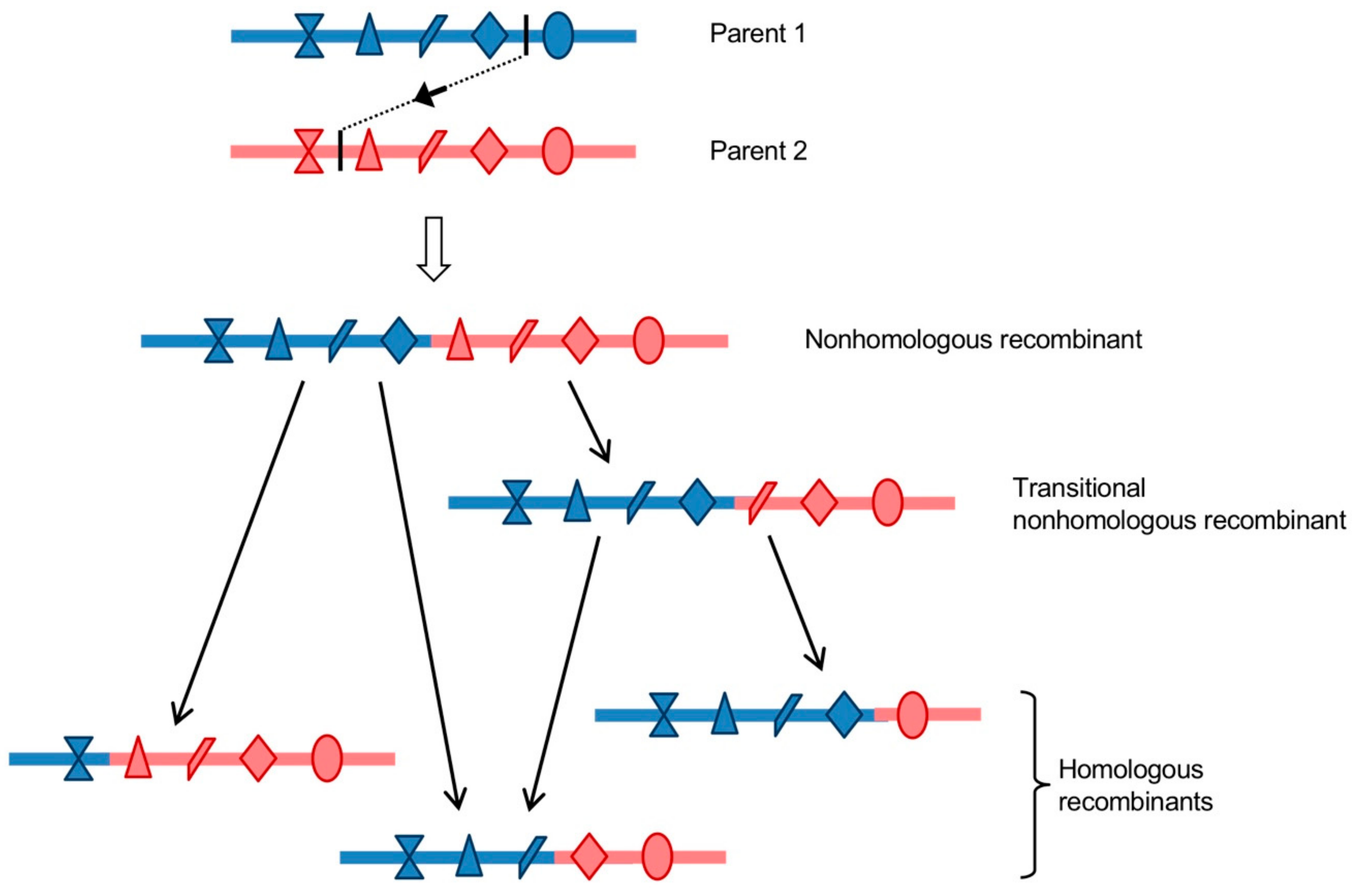
5. The Generation of Homologous Intertypic Recombinant Enteroviruses, a Multi-Step Process
6. Recombination in Enteroviruses, Experimental Evidences of a Modular Evolutionary Process
6.1. Location of Recombination Hotspots
6.2. Modular Recombination Process
7. Concluding Remarks
Funding
Conflicts of Interest
References
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef] [PubMed]
- Pallansch, M.; Roos, R. Enteroviruses: Polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2007; pp. 839–893. [Google Scholar]
- Nathanson, N.; Kew, O.M. From emergence to eradication: The epidemiology of poliomyelitis deconstructed. Am. J. Epidemiol. 2010, 172, 1213–1229. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.D.; Yergolkar, P.; Shankarappa, K.S. Antigenic diversity of enteroviruses associated with nonpolio acute flaccid paralysis, India, 2007–2009. Emerg. Infect. Dis. 2012, 18, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-T.; Huang, L.-M.; Lu, C.-Y.; Kao, C.-L.; Lee, W.-T.; Lee, P.-I.; Chen, C.-M.; Huang, F.-Y.; Lee, C.-Y.; Chang, L.-Y. Clinical features and factors of unfavorable outcomes for non-polio enterovirus infection of the central nervous system in northern Taiwan, 1994–2003. J. Microbiol. Immunol. Infect. 2005, 38, 417–424. [Google Scholar] [PubMed]
- Rudolph, H.; Schroten, H.; Tenenbaum, T. Enterovirus Infections of the Central Nervous System in Children: An Update. Pediatr. Infect. Dis. J. 2016, 35, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Tracy, S.; Chapman, N.M. Group B coxsackievirus disease. In The Picornaviruses; Ehrenfeld, E., Domingo, E., Roos, R.P., Eds.; ASM Press: Washington, DC, USA, 2010; pp. 353–368. [Google Scholar]
- Muehlenbachs, A.; Bhatnagar, J.; Zaki, S.R. Tissue tropism, pathology and pathogenesis of enterovirus infection. J. Pathol. 2015, 235, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Pons-Salort, M.; Parker, E.P.K.; Grassly, N.C. The epidemiology of non-polio enteroviruses: Recent advances and outstanding questions. Curr. Opin. Infect. Dis. 2015, 28, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Tapparel, C.; Siegrist, F.; Petty, T.J.; Kaiser, L. Picornavirus and enterovirus diversity with associated human diseases. Infect. Genet. Evol. 2013, 14, 282–293. [Google Scholar] [CrossRef]
- Jacques, J.; Moret, H.; Minette, D.; Lévêque, N.; Jovenin, N.; Deslée, G.; Lebargy, F.; Motte, J.; Andréoletti, L. Epidemiological, molecular, and clinical features of enterovirus respiratory infections in French children between 1999 and 2005. J. Clin. Microbiol. 2008, 46, 206–213. [Google Scholar] [CrossRef]
- Racaniello, V.R. Picornaviridae: The viruses and their replication. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2007; pp. 795–838. [Google Scholar]
- Lulla, V.; Dinan, A.M.; Hosmillo, M.; Chaudhry, Y.; Sherry, L.; Irigoyen, N.; Nayak, K.M.; Stonehouse, N.J.; Zilbauer, M.; Goodfellow, I.; et al. An upstream protein-coding region in enteroviruses modulates virus infection in gut epithelial cells. Nat. Microbiol. 2018, 4, 280. [Google Scholar]
- Andino, R.; Rieckhof, G.E.; Baltimore, D. A functional ribonucleoprotein complex forms around the 5′ end of poliovirus RNA. Cell 1990, 63, 369–380. [Google Scholar] [CrossRef]
- Barton, D.J.; O’Donnell, B.J.; Flanegan, J.B. 5′ cloverleaf in poliovirus RNA is a cis-acting replication element required for negative-strand synthesis. EMBO J. 2001, 20, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Rieder, E.; Xiang, W.; Paul, A.; Wimmer, E. Analysis of the cloverleaf element in a human rhinovirus type 14/poliovirus chimera: Correlation of subdomain D structure, ternary protein complex formation and virus replication. J. Gen. Virol. 2003, 84, 2203–2216. [Google Scholar] [CrossRef] [PubMed]
- Vogt, D.A.; Andino, R. An RNA element at the 5′-end of the poliovirus genome functions as a general promoter for RNA synthesis. PLoS Pathog. 2010, 6, 1000936. [Google Scholar] [CrossRef]
- Nicholson, R.; Pelletier, J.; Le, S.Y.; Sonenberg, N. Structural and functional analysis of the ribosome landing pad of poliovirus type 2: In Vivo translation studies. J. Virol. 1991, 65, 5886–5894. [Google Scholar]
- Sweeney, T.R.; Abaeva, I.S.; Pestova, T.V.; Hellen, C.U.T. The mechanism of translation initiation on type 1 picornavirus IRESs. EMBO J. 2014, 33, 76–92. [Google Scholar] [CrossRef]
- Wimmer, E.; Paul, A. The making of a Picornavirus genome. In The Picornaviruses; Ehrenfeld, E., Domingo, E., Roos, R.P., Eds.; ASM Press: Washington, DC, USA, 2010; pp. 33–55. [Google Scholar]
- Mueller, S.; Wimmer, E.; Cello, J. Poliovirus and poliomyelitis: A tale of guts, brains, and an accidental event. Virus Res. 2005, 111, 175–193. [Google Scholar] [CrossRef]
- Domingo, E.; Sheldon, J.; Perales, C. Viral Quasispecies Evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [Green Version]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, 1–8. [Google Scholar] [CrossRef]
- Pfeiffer, J.K.; Kirkegaard, K. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog. 2005, 1, 0102–0110. [Google Scholar] [CrossRef]
- Gnädig, N.F.; Beaucourt, S.; Campagnola, G.; Bordería, A.V.; Sanz-Ramos, M.; Gong, P.; Blanc, H.; Peersen, O.B.; Vignuzzi, M. Coxsackievirus B3 mutator strains are attenuated in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, E2294–E2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, T.; Kwang, J. Attenuation of Human Enterovirus 71 High-Replication-Fidelity Variants in AG129 Mice. J. Virol. 2014, 88, 5803–5815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Galli, A.; Bukh, J. Comparative analysis of the molecular mechanisms of recombination in hepatitis C virus. Trends Microbiol. 2014, 22, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Palero, F.; González-Candelas, F. Recombination in viruses: Mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Sztuba-Solińska, J.; Urbanowicz, A.; Figlerowicz, M.; Bujarski, J.J. RNA-RNA Recombination in Plant Virus Replication and Evolution. Annu. Rev. Phytopathol. 2011, 49, 415–443. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Rossolillo, P.; Negroni, M. RNA structures, genomic organization and selection of recombinant HIV. RNA Biol. 2011, 8, 37–41. [Google Scholar] [CrossRef]
- Barr, J.N.; Fearns, R. How RNA viruses maintain their genome integrity. J. Gen. Virol. 2010, 91, 1373–1387. [Google Scholar] [CrossRef]
- Rawson, J.M.O.; Nikolaitchik, O.A.; Keele, B.F.; Pathak, V.K.; Hu, W.-S. Recombination is required for efficient HIV-1 replication and the maintenance of viral genome integrity. Nucleic Acids Res. 2018, 46, 10535–10545. [Google Scholar] [CrossRef] [Green Version]
- Kempf, B.J.; Watkins, C.L.; Peersen, O.B.; Barton, D.J. Picornavirus RNA Recombination Counteracts Error Catastrophe. J. Virol. 2019, 93, 652. [Google Scholar] [CrossRef]
- Xiao, Y.; Rouzine, I.M.; Bianco, S.; Acevedo, A.; Goldstein, E.F.; Farkov, M.; Brodsky, L.; Andino, R. RNA recombination enhances adaptability and is required for virus spread and virulence. Cell Host Microbe 2016, 19, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Dolan, P.T.; Goldstein, E.F.; Li, M.; Farkov, M.; Brodsky, L.; Andino, R. Poliovirus intrahost evolution is required to overcome tissue-specific innate immune responses. Nat. Commun. 2017, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.S.; Lustig, S.; Strauss, E.G.; Strauss, J.H. Western equine encephalitis virus is a recombinant virus. Proc. Natl. Acad. Sci. USA 1988, 85, 5997–6001. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Kang, W.; Shirako, Y.; Rumenapf, T.; Strauss, E.G.; Strauss, J.H. Recombinational history and molecular evolution of western equine encephalomyelitis complex alphaviruses. J. Virol. 1997, 71, 613–623. [Google Scholar] [PubMed]
- Simmonds, P.; Midgley, S. Recombination in the genesis and evolution of hepatitis B virus genotypes. J. Virol. 2005, 79, 15467–15476. [Google Scholar] [CrossRef] [PubMed]
- Charrel, R.N.; de Lamballerie, X.; Emonet, S. Phylogeny of the genus Arenavirus. Curr. Opin. Microbiol. 2008, 11, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, M.W.; Boynton, T.O.; Hilt, D.A.; McKinley, E.T.; Kissinger, J.C.; Paterson, A.H.; Robertson, J.; Lemke, C.; McCall, A.W.; Williams, S.M.; et al. Emergence of a group 3 coronavirus through recombination. Virology 2010, 398, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marais, A.; Faure, C.; Mustafayev, E.; Candresse, T. Characterization of new isolates of Apricot vein clearing-associated virus and of a new prunus-infecting virus: Evidence for recombination as a driving force in Betaflexiviridae evolution. PLoS ONE 2015, 10, 1–15. [Google Scholar] [CrossRef]
- Kelly, A.G.; Netzler, N.E.; White, P.A. Ancient recombination events and the origins of hepatitis E virus. BMC Evol. Biol. 2016, 16, 1–18. [Google Scholar] [CrossRef]
- Miras, M.; Sempere, R.N.; Kraft, J.J.; Miller, W.A.; Aranda, M.A.; Truniger, V. Interfamilial recombination between viruses led to acquisition of a novel translation-enhancing RNA element that allows resistance breaking. New Phytol. 2014, 202, 233–246. [Google Scholar] [CrossRef]
- Shang, P.; Misra, S.; Hause, B.; Fang, Y. A naturally occurring recombinant enterovirus expresses a torovirus deubiquitinase. J. Virol. 2017, 91, JVI.00450-17. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiaka, S.; Naoi, Y.; Imai, R.; Masuda, T.; Ito, M.; Akagami, M.; Ouchi, Y.; Ishii, K.; Sakaguchi, S.; Omatsu, T.; et al. Genetic diversity and recombination of enterovirus G strains in Japanese pigs: High prevalence of strains carrying a papain-like cysteine protease sequence in the enterovirus G population. PLoS ONE 2018, 13, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.J.; Weiller, G.F. Evidence that a plant virus switched hosts to infect a vertebrate and then recombined with a vertebrate-infecting virus. Proc. Natl. Acad. Sci. USA 1999, 96, 8022–8027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alamgir, A.S.M.; Owens, N.; Lavignon, M.; Malik, F.; Evans, L.H. Precise identification of endogenous proviruses of NFS/N mice participating in recombination with moloney ecotropic murine leukemia virus (MuLV) to generate polytropic MuLVs. J. Virol. 2005, 79, 4664–4671. [Google Scholar] [CrossRef] [PubMed]
- Etienne, L.; Hahn, B.H.; Sharp, P.M.; Matsen, F.A.; Emerman, M. Gene loss and adaptation to hominids underlie the ancient origin of HIV-1. Cell Host Microbe 2013, 14, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Bedford, T. Modern-day SIV viral diversity generated by extensive recombination and cross-species transmission. PLoS Pathog. 2017, 13, 1–21. [Google Scholar] [CrossRef]
- Filomatori, C.V.; Bardossy, E.S.; Merwaiss, F.; Suzuki, Y.; Henrion, A.; Saleh, M.C.; Alvarez, D.E. RNA recombination at Chikungunya virus 3′UTR as an evolutionary mechanism that provides adaptability. PLoS Pathog. 2019, 15, 1007706. [Google Scholar] [CrossRef]
- Becher, P.; Tautz, N. RNA recombination in pestiviruses—Cellular RNA sequences in viral genomes highlight the role of host factors for viral persistence and lethal disease. RNA Biol. 2011, 8, 37–41. [Google Scholar] [CrossRef]
- Khatchikian, D.; Orlich, M.; Rott, R. Increased viral pathogenicity after insertion of a 28S ribosomal RNA sequence into the haemagglutinin gene of an influenza virus. Nature 1989, 340, 156–157. [Google Scholar] [CrossRef]
- Herrewegh, A.A.; Vennema, H.; Horzinek, M.C.; Rottier, P.J.M.; De Groot, R.J. The molecular genetics of feline coronaviruses: Comparative sequence analysis of the ORF7a/7b transcription unit of different biotypes. Virology 1995, 212, 622–631. [Google Scholar] [CrossRef]
- Fernández-Cuartero, B.; Burgyán, J.; Aranda, M.A.; Salänki, K.; Moriones, E.; Garcıa-Arenal, F. Increase in the Relative Fitness of a Plant Virus RNA Associated with Its Recombinant Nature. Virology 1994, 203, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Pilipenko, E.V.; Gmyl, A.P.; Maslova, S.V.; Svitkin, Y.V.; Sinyakov, A.N.; Agol, V.I. Prokaryotic-like cis elements in the cap-independent internal initiation of translation on picornavirus RNA. Cell 1992, 68, 119–131. [Google Scholar] [CrossRef]
- Chen, Y.; Goldbach, R.; Prins, M. Inter- and Intramolecular Recombinations in the Cucumber Mosaic Virus Genome Related to Adaptation to Alstroemeria. J. Virol. 2002, 76, 4119–4124. [Google Scholar] [CrossRef] [PubMed]
- Erickson, A.K.; Jesudhasan, P.R.; Mayer, M.J.; Narbad, A.; Winter, S.E.; Pfeiffer, J.K. Bacteria Facilitate Enteric Virus Co-infection of Mammalian Cells and Promote Genetic Recombination. Cell Host Microbe 2018, 23, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, D.J.; Minor, P.D. Vaccine Strategies. In The Picornaviruses; Ehrenfeld, E., Dominog, E., Roos, R.P., Eds.; ASM Press: Washington, DC, USA, 2010; pp. 431–447. [Google Scholar]
- Kew, O.M.; Sutter, R.W.; de Gourville, E.M.; Dowdle, W.R.; Pallansch, M.A. Vaccine-Derived Polioviruses and the Endgame Strategy for Global Polio Eradication. Annu. Rev. Microbiol. 2005, 59, 587–635. [Google Scholar] [CrossRef]
- Combelas, N.; Holmblat, B.; Joffret, M.L.; Colbère-Garapin, F.; Delpeyroux, F. Recombination between poliovirus and coxsackie A viruses of species C: A model of viral genetic plasticity and emergence. Viruses 2011, 3, 1460–1484. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.C.; Diop, O.M.; Sutter, R.W.; Kew, O.M. Vaccine-derived polioviruses. J. Infect. Dis. 2014, 210, S283–S293. [Google Scholar] [CrossRef]
- Pons-Salort, M.; Burns, C.C.; Lyons, H.; Blake, I.M.; Jafari, H.; Oberste, M.S.; Kew, O.M.; Grassly, N.C. Preventing Vaccine-Derived Poliovirus Emergence during the Polio Endgame. PLoS Pathog. 2016, 12, 1–18. [Google Scholar] [CrossRef]
- WHO Circulating Vaccine-Derived Polioviruses. Available online: http://polioeradication.org/polio-today/polio-now/this-week/circulating-vaccine-derived-poliovirus/ (accessed on 7 May 2019).
- Rakoto-Andrianarivelo, M.; Guillot, S.; Iber, J.; Balanant, J.; Blondel, B.; Riquet, F.; Martin, J.; Kew, O.; Randriamanalina, B.; Razafinimpiasa, L.; et al. Co-circulation and evolution of polioviruses and species C enteroviruses in a district of Madagascar. PLoS Pathog. 2007, 3, 1950–1961. [Google Scholar] [CrossRef]
- Rakoto-Andrianarivelo, M.; Gumede, N.; Jegouic, S.; Balanant, J.; Andriamamonjy, S.N.; Rabemanantsoa, S.; Birmingham, M.; Randriamanalina, B.; Nkolomoni, L.; Venter, M.; et al. Reemergence of Recombinant Vaccine-Derived Poliovirus Outbreak in Madagascar. J. Infect. Dis. 2008, 197, 1427–1435. [Google Scholar] [CrossRef]
- Dedepsidis, E.; Kyriakopoulou, Z.; Pliaka, V.; Kottaridi, C.; Bolanaki, E.; Levidiotou-Stefanou, S.; Komiotis, D.; Markoulatos, P. Retrospective characterization of a vaccine-derived poliovirus type 1 isolate from sewage in Greece. Appl. Environ. Microbiol. 2007, 73, 6697–6704. [Google Scholar] [CrossRef] [PubMed]
- Kew, O.; Morris-Glasgow, V.; Landaverde, M.; Burns, C.; Shaw, J.; Garib, Z.; André, J.; Blackman, E.; Freeman, C.J.; Jorba, J.; et al. Outbreak of poliomyelitis in hispaniola associated with circulating type 1 vaccine-derived poliovirus. Science 2002, 296, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Thorley, B.; Paladin, F.J.; Brussen, K.A.; Stambos, V.; Yuen, L.; Utama, A.; Tano, Y.; Arita, M.; Yoshida, H.; et al. Circulation of Type 1 Vaccine-Derived Poliovirus in the Philippines in 2001. J. Virol. 2004, 78, 13512–13521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joffret, M.L.; Jégouic, S.; Bessaud, M.; Balanant, J.; Tran, C.; Caro, V.; Holmblat, B.; Razafindratsimandresy, R.; Reynes, J.M.; Rakoto-Andrianarivelo, M.; et al. Common and diverse features of cocirculating type 2 and 3 recombinant vaccine-derived polioviruses isolated from patients with poliomyelitis and healthy children. J. Infect. Dis. 2012, 205, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Adu, F.; Iber, J.; Bukbuk, D.; Gumede, N.; Yang, S.J.; Jorba, J.; Campagnoli, R.; Sule, W.F.; Yang, C.F.; Burns, C.; et al. Isolation of recombinant type 2 vaccine-derived poliovirus (VDPV) from a Nigerian child. Virus Res. 2007, 127, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.C.; Shaw, J.; Jorba, J.; Bukbuk, D.; Adu, F.; Gumede, N.; Pate, M.A.; Abanida, E.A.; Gasasira, A.; Iber, J.; et al. Multiple Independent Emergences of Type 2 Vaccine-Derived Polioviruses during a Large Outbreak in Northern Nigeria. J. Virol. 2013, 87, 4907–4922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.-F.; Naguib, T.; Yang, S.-J.; Nasr, E.; Jorba, J.; Ahmed, N.; Campagnoli, R.; van der Avoort, H.; Shimizu, H.; Yoneyama, T.; et al. Circulation of endemic type 2 vaccine-derived poliovirus in Egypt from 1983 to 1993. J. Virol. 2003, 77, 8366–8377. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Faase, J.A.J.; Toyoda, H.; Paul, A.; Wimmer, E.; Gorbalenya, A.E. Evidence for emergence of diverse polioviruses from C-cluster coxsackie A viruses and implications for global poliovirus eradication. Proc. Natl. Acad. Sci. USA 2007, 104, 9457–9462. [Google Scholar] [CrossRef] [Green Version]
- Riquet, F.B.; Blanchard, C.; Jegouic, S.; Balanant, J.; Guillot, S.; Vibet, M.-A.; Rakoto-Andrianarivelo, M.; Delpeyroux, F. Impact of exogenous sequences on the characteristics of an epidemic type 2 recombinant vaccine-derived poliovirus. J. Virol. 2008, 82, 8927–8932. [Google Scholar] [CrossRef]
- Jegouic, S.; Joffret, M.L.; Blanchard, C.; Riquet, F.B.; Perret, C.; Pelletier, I.; Colbere-Garapin, F.; Rakoto-Andrianarivelo, M.; Delpeyroux, F. Recombination between polioviruses and co-circulating coxsackie A viruses: Role in the emergence of pathogenic vaccine-derived polioviruses. PLoS Pathog. 2009, 5, 1000412. [Google Scholar] [CrossRef]
- Muslin, C.; Joffret, M.-L.; Pelletier, I.; Blondel, B.; Delpeyroux, F. Evolution and Emergence of Enteroviruses through Intra- and Inter-species Recombination: Plasticity and Phenotypic Impact of Modular Genetic Exchanges in the 5′ Untranslated Region. PLoS Pathog. 2015, 11, 1005266. [Google Scholar] [CrossRef] [PubMed]
- Bessaud, M.; Joffret, M.L.; Blondel, B.; Delpeyroux, F. Exchanges of genomic domains between poliovirus and other cocirculating species C enteroviruses reveal a high degree of plasticity. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, K.; Baltimore, D. The mechanism of RNA recombination in poliovirus. Cell 1986, 47, 433–443. [Google Scholar] [CrossRef]
- Oprisan, G.; Combiescu, M.; Guillot, S.; Caro, V.; Combiescu, A.; Delpeyroux, F.; Crainic, R. Natural genetic recombination between co-circulating heterotypic enteroviruses. J. Gen. Virol. 2002, 83, 2193–2200. [Google Scholar] [CrossRef] [PubMed]
- Chevaliez, S.; Szendröi, A.; Caro, V.; Balanant, J.; Guillot, S.; Berencsi, G.; Delpeyroux, F. Molecular comparison of echovirus 11 strains circulating in Europe during an epidemic of multisystem hemorrhagic disease of infants indicates that evolution generally occurs by recombination. Virology 2004, 325, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, M.D.; Volle, R.; Joffret, M.L.; Sadeuh-Mba, S.A.; Gouandjika-Vasilache, I.; Kebe, O.; Wiley, M.R.; Majumdar, M.; Simon-Loriere, E.; Sakuntabhai, A.; et al. Genetic characterization of enterovirus A71 circulating in Africa. Emerg. Infect. Dis. 2018, 24, 754–757. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, A.N.; Lashkevich, V.A.; Ivanova, O.E.; Koroleva, G.A.; Hinkkanen, A.E.; Ilonen, J. Recombination in circulating enteroviruses. J. Virol. 2003, 77, 10423–10431. [Google Scholar] [CrossRef]
- McWilliam Leitch, E.C.; Cabrerizo, M.; Cardosa, J.; Harvala, H.; Ivanova, O.E.; Koike, S.; Kroes, A.C.M.; Lukashev, A.; Perera, D.; Roivainen, M.; et al. The Association of Recombination Events in the Founding and Emergence of Subgenogroup Evolutionary Lineages of Human Enterovirus 71. J. Virol. 2012, 86, 2676–2685. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, A.M.; Andersson, P.; Savolainen, C.; Mulders, M.N.; Hovi, T. Evolution of the genome of Human enterovirus B: Incongruence between phylogenies of the VP1 and 3CD regions indicates frequent recombination within the species. J. Gen. Virol. 2003, 84, 1223–1235. [Google Scholar] [CrossRef]
- Bessaud, M.; Joffret, M.L.; Holmblat, B.; Razafindratsimandresy, R.; Delpeyroux, F. Genetic relationship between cocirculating human enteroviruses species C. PLoS ONE 2011, 6, e24823. [Google Scholar] [CrossRef]
- Kyriakopoulou, Z.; Pliaka, V.; Amoutzias, G.D.; Markoulatos, P. Recombination among human non-polio enteroviruses: Implications for epidemiology and evolution. Virus Genes 2015, 50, 177–188. [Google Scholar] [CrossRef]
- Tan, Y.; Hassan, F.; Schuster, J.E.; Simenauer, A.; Selvarangan, R.; Halpin, R.A.; Lin, X.; Fedorova, N.; Stockwell, T.B.; Lam, T.T.-Y.; et al. Molecular Evolution and Intraclade Recombination of Enterovirus D68 during the 2014 Outbreak in the United States. J. Virol. 2016, 90, 1997–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Zhang, Y.; Yan, D.; Zhu, S.; Wang, D.; Ji, T.; Li, X.; Song, Y.; Gu, X.; Xu, W. Two genotypes of coxsackievirus A2 associated with hand, foot, and mouth disease circulating in China since 2008. PLoS ONE 2016, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hassel, C.; Mirand, A.; Farkas, A.; Diedrich, S.; Huemer, H.P.; Peigue-Lafeuille, H.; Archimbaud, C.; Henquell, C.; Bailly, J.-L.; Network, H.F.S. Phylogeography of Coxsackievirus A16 Reveals Global Transmission Pathways and Recent Emergence and Spread of a Recombinant Genogroup. J. Virol. 2017, 91, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Holmes, E.C. Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 1999, 80, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Shukla, D.; Srivastava, S.; Idris, M.Z.; Dhole, T.N. High frequency of enterovirus serotype circulation in a densely populated area of India. J. Infect. Dev. Ctries. 2013, 7, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Sadeuh-Mba, S.A.; Bessaud, M.; Massenet, D.; Joffret, M.L.; Endegue, M.C.; Njouom, R.; Reynes, J.M.; Rousset, D.; Delpeyroux, F. High frequency and diversity of species C enteroviruses in Cameroon and neighboring countries. J. Clin. Microbiol. 2013, 51, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Thoelen, I.; Lemey, P.; Van der Donck, I.; Beuselinck, K.; Lindberg, A.M.; Van Ranst, M. Molecular typing and epidemiology of enteroviruses identified from an outbreak of aseptic meningitis in Belgium during the summer of 2000. J. Med. Virol. 2003, 70, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Volle, R.; Bailly, J.-L.; Mirand, A.; Pereira, B.; Marque-Juillet, S.; Chambon, M.; Regagnon, C.; Brebion, A.; Henquell, C.; Peigue-Lafeuille, H.; et al. Variations in Cerebrospinal Fluid Viral Loads Among Enterovirus Genotypes in Patients Hospitalized With Laboratory-Confirmed Meningitis Due to Enterovirus. J. Infect. Dis. 2014, 210, 576–584. [Google Scholar] [CrossRef] [Green Version]
- Mirand, A.; Henquell, C.; Archimbaud, C.; Chambon, M.; Charbonne, F.; Peigue-Lafeuille, H.; Bailly, J.L. Prospective identification of enteroviruses involved in meningitis in 2006 through direct genotyping in cerebrospinal fluid. J. Clin. Microbiol. 2008, 46, 87–96. [Google Scholar] [CrossRef]
- Volle, R.; Razafindratsimandresy, R.; Joffret, M.L.; Bessaud, M.; Rabemanantsoa, S.; Andriamamonjy, S.; Raharinantoanina, J.; Blondel, B.; Heraud, J.-M.; Bailly, J.-L.; et al. High permissiveness for genetic exchanges between enteroviruses of species A, including enterovirus 71, favours evolution through intertypic recombination in Madagascar. J. Virol. 2019, 93, e01667-18. [Google Scholar] [CrossRef] [PubMed]
- Han, J.F.; Zhang, Y.; Hou, P.Q.; Zhu, S.Y.; Wu, X.Y.; Zhao, H.; Yu, M.; Qin, C.F. Human enterovirus co-infection in severe HFMD patients in China. J. Clin. Virol. 2014, 61, 621–622. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, E.R.; Erickson, A.K.; Jesudhasan, P.R.; Robinson, C.M.; Pfeiffer, J.K. Plaques formed by mutagenized viral populations have elevated coinfection frequencies. MBio 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A. Phosphatidylserine Vesicles Enable Efficient En Bloc Transmission of Enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egger, D.; Bienz, K. Recombination of poliovirus RNA proceeds in mixed replication complexes originating from distinct replication start sites. J. Virol. 2002, 76, 10960–10971. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, H.; Diaz, A.; Ahlquist, P. Intermolecular RNA Recombination Occurs at Different Frequencies in Alternate Forms of Brome Mosaic Virus RNA Replication Compartments. Viruses 2018, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Nair, V.; Hansen, B.T.; Hoyt, F.H.; Fischer, E.R.; Ehrenfeld, E. Complex Dynamic Development of Poliovirus Membranous Replication Complexes. J. Virol. 2012, 86, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santti, J.; Hyypiä, T.; Kinnunen, L.; Salminen, M. Evidence of recombination among enteroviruses. J. Virol. 1999, 73, 8741–8749. [Google Scholar] [PubMed]
- Brown, B.; Oberste, M.S.; Maher, K.; Mark, A.; Pallansch, M.A. Complete Genomic Sequencing Shows that Polioviruses and Members of Human Enterovirus Species C Are Closely Related in the Noncapsid Coding Region. J. Virol. 2003, 77, 8973–8984. [Google Scholar] [CrossRef] [Green Version]
- Lukashev, A.N. Role of recombination in evolution of enteroviruses. Rev. Med. Virol. 2005, 15, 157–167. [Google Scholar] [CrossRef]
- Oberste, M.S.; Peñaranda, S.; Pallansch, M.A. RNA recombination plays a major role in genomic change during circulation of coxsackie B viruses. J. Virol. 2004, 78, 2948–2955. [Google Scholar] [CrossRef] [PubMed]
- Nikolaidis, M.; Mimouli, K.; Kyriakopoulou, Z.; Tsimpidis, M.; Tsakogiannis, D.; Markoulatos, P.; Amoutzias, G.D. Large-scale genomic analysis reveals recurrent patterns of intertypic recombination in human enteroviruses. Virology 2019, 526, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Oberste, M.S.; Peñaranda, S.; Maher, K.; Pallansch, M.A. Complete genome sequences of all members of the species Human enterovirus A. J. Gen. Virol. 2004, 85, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Oberste, M.S.; Maher, K.; Pallansch, M.A. Evidence for frequent recombination within species human enterovirus B based on complete genomic sequences of all thirty-seven serotypes. J. Virol. 2004, 78, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Welch, J. Frequency and Dynamics of Recombination within Different Species of Human Enteroviruses. J. Virol. 2006, 80, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Bouslama, L.; Nasri, D.; Chollet, L.; Belguith, K.; Bourlet, T.; Aouni, M.; Pozzetto, B.; Pillet, S. Natural recombination event within the capsid genomic region leading to a chimeric strain of human enterovirus B. J. Virol. 2007, 81, 8944–8952. [Google Scholar] [CrossRef]
- Martín, J.; Samoilovich, E.; Dunn, G.; Lackenby, A.; Feldman, E.; Heath, A.; Svirchevskaya, E.; Cooper, G.; Yermalovich, M.; Minor, P.D. Isolation of an Intertypic Poliovirus Capsid Recombinant from a Child with Vaccine-Associated Paralytic Poliomyelitis. J. Virol. 2002, 76, 10921–10928. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Huang, K.; Li, L.; Wu, X.; Zheng, L.; Wan, C.; Zhao, W.; Ke, C.; Zhang, B. Complete genome sequence analysis of human echovirus 30 isolated during a large outbreak in Guangdong Province of China, in 2012. Arch. Virol. 2014, 159, 379–383. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, S.; Yan, D.; Liu, G.; Bai, R.; Wang, D.; Chen, L.; Zhu, H.; An, H.; Kew, O.; et al. Natural type 3/type 2 intertypic vaccine-related poliovirus recombinants with the first crossover sites within the VP1 capsid coding region. PLoS ONE 2010, 5, 1–11. [Google Scholar] [CrossRef]
- Blomqvist, S.; Bruu, A.-L.; Stenvik, M.; Hovi, T. Characterization of a recombinant type 3/type 2 poliovirus isolated from a healthy vaccinee and containing a chimeric capsid protein VP1. J. Gen. Virol. 2003, 84, 573–580. [Google Scholar] [CrossRef]
- Korotkova, E.; Laassri, M.; Zagorodnyaya, T.; Petrovskaya, S.; Rodionova, E.; Cherkasova, E.; Gmyl, A.; Ivanova, O.E.; Eremeeva, T.P.; Lipskaya, G.Y.; et al. Pressure for pattern-specific intertypic recombination between sabin polioviruses: Evolutionary implications. Viruses 2017, 9, 353. [Google Scholar] [CrossRef]
- Lukashev, A.N.; Elena, Y.; Belalov, I.S.; Ivanova, O.E.; Eremeeva, T.P.; Reznik, V.I.; Trotsenko, O.E.; Drexler, J.F.; Drosten, C. Recombination strategies and evolutionary dynamics of the Human enterovirus A global gene pool. J. Gen. Virol. 2014, 95, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Smura, T.; Blomqvist, S.; Vuorinen, T.; Ivanova, O.; Samoilovich, E.; Al-Hello, H.; Savolainen-Kopra, C.; Hovi, T.; Roivainen, M. Recombination in the evolution of enterovirus C species sub-group that contains types CVA-21, CVA-24, EV-C95, EV-C96 and EV-C99. PLoS ONE 2014, 9, e94579. [Google Scholar] [CrossRef]
- Arita, M.; Zhu, S.-L.; Yoshida, H.; Yoneyama, T.; Miyamura, T.; Shimizu, H. A Sabin 3-Derived Poliovirus Recombinant Contained a Sequence Homologous with Indigenous Human Enterovirus Species C in the Viral Polymerase Coding Region. J. Virol. 2005, 79, 12650–12657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowry, K.; Woodman, A.; Cook, J.; Evans, D.J. Recombination in Enteroviruses is a Biphasic Replicative Process Involving the Generation of Greater-than Genome Length “Imprecise” Intermediates. PLoS Pathog. 2014, 10, 1004191. [Google Scholar] [CrossRef] [PubMed]
- Holmblat, B.; Jégouic, S.; Muslin, C.; Blondel, B.; Joffret, M.-L.; Delpeyroux, F. Nonhomologous recombination between defective poliovirus and coxsackievirus genomes suggests a new model of genetic plasticity for picornaviruses. MBio 2014, 5, e01119-14. [Google Scholar] [CrossRef]
- McWilliam Leitch, E.C.; Bendig, J.; Cabrerizo, M.; Cardosa, J.; Hyypia, T.; Ivanova, O.E.; Kelly, A.; Kroes, A.C.M.; Lukashev, A.; MacAdam, A.; et al. Transmission Networks and Population Turnover of Echovirus 30. J. Virol. 2009, 83, 2109–2118. [Google Scholar] [CrossRef] [Green Version]
- Gmyl, A.P.; Belousov, E.V.; Maslova, S.V.; Khitrina, E.V.; Chetverin, A.B.; Agol, V.I. Nonreplicative RNA recombination in poliovirus. J. Virol. 1999, 73, 8958–8965. [Google Scholar] [PubMed]
- Nagy, P.D.; Simon, A.E. New Insights into the Mechanisms of RNA Recombination. Virology 1997, 235, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Cooper, P.D.; Steiner-Pryor, A.; Scotti, P.D.; Delong, D. On the Nature of Poliovirus Genetic Recombinants. J. Gen. Virol. 1974, 23, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Galetto, R.; Negroni, M. Mechanistic features of recombination in HIV. AIDS Rev. 2005, 7, 92–102. [Google Scholar] [PubMed]
- Lai, M.M. RNA recombination in animal and plant viruses. Microbiol. Rev. 1992, 56, 61–79. [Google Scholar] [PubMed]
- Arnold, J.J.; Cameron, C.E. Poliovirus RNA-dependent RNA Polymerase (3D pol) Is Sufficient for Template Switching in Vitro. J. Biol. Chem. 1999, 274, 2706–2716. [Google Scholar] [CrossRef] [PubMed]
- Flanegan, J.B.; Baltimore, D. Poliovirus-specific primer-dependent RNA polymerase able to copy poly(A). Proc. Natl. Acad. Sci. USA 1977, 74, 3677–3680. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.W.; Richards, O.C.; Dmitrieva, T.M.; Agol, V.; Ehrenfeld, E. RNA duplex unwinding activity of poliovirus RNA-dependent RNA polymerase 3D(pol). J. Virol. 1993, 67, 3010–3018. [Google Scholar] [PubMed]
- Chetverin, A.B.; Kopein, D.S.; Chetverina, H.V.; Demidenko, A.A.; Ugarov, V.I. Viral RNA-directed RNA polymerases use diverse mechanisms to promote recombination between RNA molecules. J. Biol. Chem. 2005, 280, 8748–8755. [Google Scholar] [CrossRef] [PubMed]
- Woodman, A.; Arnold, J.J.; Cameron, C.E.; Evans, D.J. Biochemical and genetic analysis of the role of the viral polymerase in enterovirus recombination. Nucleic Acids Res. 2016, 44, 6883–6895. [Google Scholar] [CrossRef]
- Kempf, B.J.; Peersen, O.B.; Barton, D.J. Poliovirus Polymerase Leu420 Facilitates RNA Recombination and Ribavirin Resistance. J. Virol. 2016, 90, 8410–8421. [Google Scholar] [CrossRef] [Green Version]
- Gmyl, A.P.; Agol, V.I. Diverse mechanisms of RNA recombination. Mol. Biol. 2005, 39, 529–542. [Google Scholar] [CrossRef]
- Kuge, S.; Saito, I.; Nomoto, A. Primary structure of poliovirus defective-interfering particle genomes and possible generation mechanisms of the particles. J. Mol. Biol. 1986, 192, 473–487. [Google Scholar] [CrossRef]
- Li, Y.; Ball, L. A Nonhomologous RNA recombination during negative-strand synthesis of flock house virus RNA. J. Virol. 1993, 67, 3854–3860. [Google Scholar] [PubMed]
- King, A.M. Preferred sites of recombination in poliovirus RNA: An analysis of 40 intertypic cross-over sequences. Nucleic Acids Res. 1988, 16, 11705–11723. [Google Scholar] [CrossRef] [PubMed]
- Cascone, P.J.; Carpenter, C.D.; Li, X.H.; Simon, A.E. Recombination between satellite and genomic RNAs of turnip crinkle virus. Virology 1990, 184, 791–794. [Google Scholar]
- Carpenter, C.D.; Oh, J.; Zhang, C.; Simon, A.E. Involvement of a Stem-loop Structure in the Location of Junction Sites in Viral RNA Recombination. J. Mol. Biol. 1995, 245, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Dzianott, A.; Rauffer-Bruyere, N.; Bujarski, J.J. Studies on functional interaction between brome mosaic virus replicase proteins during RNA recombination, using combined mutants in vivo and in vitro. Virology 2001, 289, 137–149. [Google Scholar] [CrossRef]
- Olsthoorn, R.C.L.; Bruyere, A.; Dzianott, A.; Bujarski, J.J. RNA recombination in brome mosaic virus: Effects of strand-specific stem-loop inserts. J. Virol. 2002, 76, 12654–12662. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, W.J.; Woods, R.J.; McCrone, J.T.; Woodman, A.; Arnold, J.J.; Yennawar, M.; Evans, R.; Cameron, C.E.; Lauring, A.S. A speed-fidelity trade-off determines the mutation rate and virulence of an RNA virus. PLoS Biol. 2018, 16, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kautz, T.F.; Forrester, N.L. RNA Virus Fidelity Mutants: A Useful Tool for Evolutionary Biology or a Complex Challenge? Viruses 2018, 10, 1–17. [Google Scholar] [CrossRef]
- Dulin, D.; Vilfan, I.D.; Berghuis, B.A.; Hage, S.; Bamford, D.H.; Poranen, M.M.; Depken, M.; Dekker, N.H. Elongation-Competent Pauses Govern the Fidelity of a Viral RNA-Dependent RNA Polymerase. Cell Rep. 2015, 10, 983–992. [Google Scholar] [CrossRef] [Green Version]
- Dulin, D.; Arnold, J.J.; van Laar, T.; Oh, H.-S.; Lee, C.; Perkins, A.L.; Harki, D.A.; Depken, M.; Cameron, C.E.; Dekker, N.H. Signatures of Nucleotide Analog Incorporation by an RNA-Dependent RNA Polymerase Revealed Using High-Throughput Magnetic Tweezers. Cell Rep. 2017, 21, 1063–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roda, R.H.; Balakrishnan, M.; Kim, J.K.; Roques, B.P.; Fay, P.J.; Bambara, R.A. Strand transfer occurs in retroviruses by a pause-initiated two-step mechanism. J. Biol. Chem. 2002, 277, 46900–46911. [Google Scholar] [CrossRef] [PubMed]
- White, K.A.; Morris, T.J. RNA determinants of junction site selection in RNA virus recombinants and defective interfering RNAs. RNA 1995, 1, 105–109. [Google Scholar]
- Gao, L.; Balakrishnan, M.; Roques, B.P.; Bambara, R.A. Insights into the multiple roles of pausing in HIV-1 reverse transcriptase-promoted strand transfers. J. Biol. Chem. 2007, 282, 6222–6231. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Martin, D.P.; Weeks, K.M.; Negroni, M. RNA Structures Facilitate Recombination-Mediated Gene Swapping in HIV-1. J. Virol. 2010, 84, 12675–12682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedepsidis, E.; Kyriakopoulou, Z.; Pliaka, V.; Markoulatos, P. Correlation between recombination junctions and RNA secondary structure elements in poliovirus Sabin strains. Virus Genes 2010, 41, 181–191. [Google Scholar] [CrossRef]
- Runckel, C.; Westesson, O.; Andino, R.; DeRisi, J.L. Identification and Manipulation of the Molecular Determinants Influencing Poliovirus Recombination. PLoS Pathog. 2013, 9, 1003164. [Google Scholar] [CrossRef]
- Tolskaya, E.A.; Romanova, L.I.; Blinov, V.M.; Viktorova, E.G.; Sinyakov, A.N.; Kolesnikova, M.S.; Agol, V.I. Studies on the recombination between RNA genomes of poliovirus: The primary structure and nonrandom distribution of crossover regions in the genomes of intertypic poliovirus recombinants. Virology 1987, 161, 54–61. [Google Scholar] [CrossRef]
- Romanova, L.I.; Blinov, V.M.; Tolskaya, E.A.; Viktorova, E.G.; Kolesnikova, M.S.; Guseva, E.A.; Agol, V.I. The primary structure of crossover regions of intertypic poliovirus recombinants: A model of recombination between RNA genomes. Virology 1986, 155, 202–213. [Google Scholar] [CrossRef]
- Bujarski, J.J.; Dzianott, A.M. Generation and analysis of nonhomologous RNA-RNA recombinants in brome mosaic virus: Sequence complementarities at crossover sites. J. Virol. 1991, 65, 4153–4159. [Google Scholar]
- Meyers, G.; Tautz, N.; Dubovi, E.J.; Thiel, H.J. Viral cytopathogenicity correlated with integration of ubiquitin-coding sequences. Virology 1991, 180, 602–616. [Google Scholar] [CrossRef]
- Nagy, P.D.; Bujarski, J.J. Targeting the site of RNA-RNA recombination in brome mosaic virus with antisense sequences. Proc. Natl. Acad. Sci. USA 1993, 90, 6390–6394. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kao, C. Factors regulating template switch in vitro by viral RNA-dependent RNA polymerases: Implications for RNA-RNA recombination. Proc. Natl. Acad. Sci. USA 2001, 98, 4972–4977. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.D.; Bujarski, J.J. Homologous RNA recombination in brome mosaic virus: AU-rich sequences decrease the accuracy of crossovers. J Virol. 1996, 70, 415–426. [Google Scholar] [PubMed]
- Nagy, P.D.; Bujarski, J.J. Engineering of homologous recombination hotspots with AU-rich sequences in brome mosaic virus. J. Virol. 1997, 71, 3799–3810. [Google Scholar] [PubMed]
- Shapka, N.; Nagy, P.D. The AU-rich RNA recombination hot spot sequence of Brome mosaic virus is functional in tombusviruses: Implications for the mechanism of RNA recombination. J. Virol. 2004, 78, 2288–2300. [Google Scholar] [CrossRef] [PubMed]
- Pilipenko, E.V.; Gmyl, A.P.; Agol, V.I. A model for rearrangements in RNA genomes. Nucleic Acids Res. 1995, 23, 1870–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallei, A.; Orlich, M.; Thiel, H.; Becher, P. Noncytopathogenic Pestivirus Strains Generated by Nonhomologous RNA Recombination: Alterations in the NS4A/NS4B Coding Region. Society 2005, 79, 14261–14270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, P.D.; Bujarski, J.J. Efficient system of homologous RNA recombination in brome mosaic virus: Sequence and structure requirements and accuracy of crossovers. J. Virol. 1995, 69, 131–140. [Google Scholar]
- Nagy, P.D. The roles of host factors in tombusvirus rna recombination. Adv. Virus Res. 2011, 81, 63–84. [Google Scholar]
- Chuang, C.; Prasanth, K.R.; Nagy, P.D. Coordinated Function of Cellular DEAD-Box Helicases in Suppression of Viral RNA Recombination and Maintenance of Viral Genome Integrity. PLoS Pathog. 2015, 11, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Prasanth, K.R.; Kovalev, N.; de Castro Martín, I.F.; Baker, J.; Nagy, P.D. Screening a yeast library of temperature-sensitive mutants reveals a role for actin in tombusvirus RNA recombination. Virology 2016, 489, 233–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Nuss, D.L. A host dicer is required for defective viral RNA production and recombinant virus vector RNA instability for a positive sense RNA virus. Proc. Natl. Acad. Sci. USA 2008, 105, 16749–16754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Choi, G.H.; Nuss, D.L. A single Argonaute gene is required for induction of RNA silencing antiviral defense and promotes viral RNA recombination. Proc. Natl. Acad. Sci. USA 2009, 106, 17927–17932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chetverin, A.B.; Chetverina, H.V.; Demidenko, A.A.; Ugarov, V.I. Nonhomologous RNA recombination in a cell-free system: Evidence for a transesterification mechanism guided by secondary structure. Cell 1997, 88, 503–513. [Google Scholar] [CrossRef]
- Chetverina, H.V.; Demidenko, A.A.; Ugarov, V.I.; Chetverin, A.B. Spontaneous rearrangements in RNA sequences. FEBS Lett. 1999, 450, 89–94. [Google Scholar] [CrossRef]
- Gmyl, A.P.; Korshenko, S.A.; Belousov, E.V.; Khitrina, E.V.; Agol, V.I. Nonreplicative homologous RNA recombination: Promiscuous joining of RNA pieces? RNA 2003, 9, 1221–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallei, A.; Pankraz, A.; Thiel, H.-J.; Becher, P. RNA Recombination In Vivo in the Absence of Viral Replication. J. Virol. 2004, 78, 6271–6281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austermann-Busch, S.; Becher, P. RNA Structural Elements Determine Frequency and Sites of Nonhomologous Recombination in an Animal Plus-Strand RNA Virus. J. Virol. 2012, 86, 7393–7402. [Google Scholar] [CrossRef] [Green Version]
- Scheel, T.K.H.; Galli, A.; Li, Y.P.; Mikkelsen, L.S.; Gottwein, J.M.; Bukh, J. Productive Homologous and Non-homologous Recombination of Hepatitis C Virus in Cell Culture. PLoS Pathog. 2013, 9, 1–12. [Google Scholar] [CrossRef]
- Kleine-Büning, M.; Meyer, D.; Austermann-Busch, S.; Roman-Sosa, G.; Rümenapf, T.; Becher, P. Nonreplicative RNA recombination of an animal plus-strand RNA virus in the absence of efficient translation of viral proteins. Genome Biol. Evol. 2017, 9, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Li, W.M.; Barnes, T.; Lee, C.H. Endoribonucleases-enzymes gaining spotlight in mRNA metabolism. FEBS J. 2010, 277, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Popow, J.; Schleiffer, A.; Martinez, J. Diversity and roles of (t)RNA ligases. Cell. Mol. Life Sci. 2012, 69, 2657–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutay, A.V.; Zenkova, M.A.; Vlassov, V.V. Nonenzymatic Recombination of RNA: Possible Mechanism for the Formation of Novel Sequences. Chem. Biodivers. 2007, 4, 762–767. [Google Scholar] [PubMed]
- Nechaev, S.; Lutay, A.; Vlassov, V.; Zenkova, M.; Nechaev, S.Y.; Lutay, A.V.; Vlassov, V.V.; Zenkova, M.A. Non-Enzymatic Template-Directed Recombination of RNAs. Int. J. Mol. Sci. 2009, 10, 1788–1807. [Google Scholar] [PubMed]
- Staroseletz, Y.; Nechaev, S.; Bichenkova, E.; Bryce, R.A.; Watson, C.; Vlassov, V.; Zenkova, M. Non-enzymatic recombination of RNA: Ligation in loops. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 705–725. [Google Scholar] [CrossRef] [Green Version]
- Mutschler, H.; Taylor, A.I.; Porebski, B.T.; Lightowlers, A.; Houlihan, G.; Abramov, M.; Herdewijn, P.; Holliger, P. Random-sequence genetic oligomer pools display an innate potential for ligation and recombination. Elife 2018, 7, 43022. [Google Scholar] [CrossRef]
- Chang, J.; Taylor, J. In vivo RNA-directed transcription, with template switching, by a mammalian RNA polymerase. EMBO J. 2002, 21, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Gudima, S.O.; Chang, J.; Taylor, J.M. Reconstitution in cultured cells of replicating HDV RNA from pairs of less than full-length RNAs. RNA 2005, 11, 90–98. [Google Scholar] [CrossRef]
- Rackwitz, H.R.; Rohde, W.; Sänger, H.L. DNA-dependent RNA polymerase II of plant origin transcribes viroid RNA into full-length copies. Nature 1981, 291, 297–301. [Google Scholar] [CrossRef]
- Schibler, M.; Piuz, I.; Hao, W.; Tapparel, C. Chimeric Rhinoviruses Obtained via Genetic Engineering or Artificially Induced Recombination Are Viable Only if the Polyprotein Coding Sequence Derives from the Same Species. J. Virol. 2015, 89, 4470–4480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodman, A.; Lee, K.-M.; Janissen, R.; Gong, Y.-N.; Dekker, N.H.; Shih, S.-R.; Cameron, C.E. Predicting Intraserotypic Recombination in Enterovirus 71. J. Virol. 2018, 93, e02057-18. [Google Scholar] [CrossRef] [PubMed]
- Holmblat, B. Etudes des Echanges Génétiques entre Poliovirus et Coxsackievirus: Un Modèle d’écosystème, d’évolution et d’émergence Virale. Ph.D. Thesis, Paris Diderot University, Paris, France, 2012. [Google Scholar]
- Kuge, S.; Nomoto, A. Construction of viable deletion and insertion mutants of the Sabin strain of type 1 poliovirus: Function of the 5′ noncoding sequence in viral replication. J. Virol. 1987, 61, 1478–1487. [Google Scholar] [PubMed]
- Gmyl, A.P.; Pilipenko, E.V.; Maslova, S.V.; Belov, G.A.; Agol, V.I. Functional and genetic plasticities of the poliovirus genome: Quasi-infectious RNAs modified in the 5′-untranslated region yield a variety of pseudorevertants. J Virol 1993, 67, 6309–6316. [Google Scholar] [PubMed]
- Baroth, M.; Orlich, M.; Thiel, H.J.; Becher, P. Insertion of cellular NEDD8 coding sequences in a pestivirus. Virology 2000, 278, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.R.; Hu, W.S.; Pathak, V.K. Relative rates of retroviral reverse transcriptase template switching during RNA- and DNA-dependent DNA synthesis. J. Virol. 1998, 72, 5198–5206. [Google Scholar] [PubMed]
- Poirier, E.Z.; Mounce, B.C.; Rozen-gagnon, K.; Hooikaas, P.J.; Stapleford, K.A.; Moratorio, G.; Vignuzzi, M. Low-Fidelity Polymerases of Alphaviruses Recombine at Higher Rates To Overproduce Defective Interfering Particles. J. Virol. 2016, 90, 2446–2454. [Google Scholar] [CrossRef] [Green Version]
- Lazzarini, R.A.; Keene, J.D.; Schubert, M. The origins of defective interfering particles of the negative-strand RNA viruses. Cell 1981, 26, 145–154. [Google Scholar] [CrossRef]
- An, W.; Telesnitsky, A. Effects of varying sequence similarity on the frequency of repeat deletion during reverse transcription of a human immunodeficiency virus type 1 vector. J. Virol. 2002, 76, 7897–7902. [Google Scholar] [CrossRef]
- Lin, C.-C.; Yang, Z.-W.; Iang, S.-B.; Chao, M. Reduced genetic distance and high replication levels increase the RNA recombination rate of hepatitis delta virus. Virus Res. 2015, 195, 79–85. [Google Scholar] [CrossRef]
- Koike, S.; Taya, C.; Aoki, J.; Matsuda, Y.; Ise, I.; Takeda, H.; Matsuzaki, T.; Amanuma, H.; Yonekawa, H.; Nomoto, A. Characterization of three different transgenic mouse lines that carry human poliovirus receptor gene—Influence of the transgene expression on pathogenesis. Arch. Virol. 1994, 139, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Nagata, N.; Iwasaki, T.; Ami, Y.; Sato, Y.; Hatano, I.; Harashima, A.; Suzaki, Y.; Yoshii, T.; Hashikawa, T.; Sata, T.; et al. A poliomyelitis model through mucosal infection in transgenic mice bearing human poliovirus receptor, TgPVR21. Virology 2004, 321, 87–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heath, L.; van der Walt, E.; Varsani, A.; Martin, D.P. Recombination Patterns in Aphthoviruses Mirror Those Found in Other Picornaviruses. J. Virol. 2006, 80, 11827–11832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twiddy, S.S.; Holmes, E.C. The extent of homologous recombination in members of the genus Flavivirus. J. Gen. Virol. 2003, 84, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Agol, V.I.; Gmyl, A.P. Viral security proteins: Counteracting host defences. Nat. Rev. Microbiol. 2010, 8, 867–878. [Google Scholar] [CrossRef]
- Teterina, N.L.; Levenson, E.A.; Ehrenfeld, E. Viable Polioviruses That Encode 2A Proteins with Fluorescent Protein Tags. J. Virol. 2010, 84, 1477–1488. [Google Scholar] [CrossRef] [Green Version]
- Sadeuh-Mba, S.A.; Bessaud, M.; Joffret, M.-L.; Endegue Zanga, M.-C.; Balanant, J.; Mpoudi Ngole, E.; Njouom, R.; Reynes, J.-M.; Delpeyroux, F.; Rousset, D. Characterization of Enteroviruses from Non-Human Primates in Cameroon Revealed Virus Types Widespread in Humans along with Candidate New Types and Species. PLoS Negl. Trop. Dis. 2014, 8, e3052. [Google Scholar] [CrossRef]
- Hellen, C.U.T.; de Breyne, S. A Distinct Group of Hepacivirus/Pestivirus-Like Internal Ribosomal Entry Sites in Members of Diverse Picornavirus Genera: Evidence for Modular Exchange of Functional Noncoding RNA Elements by Recombination. J. Virol. 2007, 81, 5850–5863. [Google Scholar] [CrossRef]
- Hu, B.; Zeng, L.P.; Yang, X.L.; Ge, X.Y.; Zhang, W.; Li, B.; Xie, J.Z.; Shen, X.R.; Zhang, Y.Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, 1–27. [Google Scholar] [CrossRef]
- Kolondam, B.; Rao, P.; Sztuba-Solinska, J.; Weber, P.H.; Dzianott, A.; Johns, M.A.; Bujarski, J.J. Co-infection with two strains of Brome mosaic bromovirus reveals common RNA recombination sites in different hosts. Virus Evol. 2015, 1, vev021. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muslin, C.; Mac Kain, A.; Bessaud, M.; Blondel, B.; Delpeyroux, F. Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process. Viruses 2019, 11, 859. https://doi.org/10.3390/v11090859
Muslin C, Mac Kain A, Bessaud M, Blondel B, Delpeyroux F. Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process. Viruses. 2019; 11(9):859. https://doi.org/10.3390/v11090859
Chicago/Turabian StyleMuslin, Claire, Alice Mac Kain, Maël Bessaud, Bruno Blondel, and Francis Delpeyroux. 2019. "Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process" Viruses 11, no. 9: 859. https://doi.org/10.3390/v11090859
APA StyleMuslin, C., Mac Kain, A., Bessaud, M., Blondel, B., & Delpeyroux, F. (2019). Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process. Viruses, 11(9), 859. https://doi.org/10.3390/v11090859