Host Immune Response Driving SARS-CoV-2 Evolution



Abstract
:1. Introduction
2. Methods and Materials
2.1. SNP Genotyping
2.2. Multiple Sequence Alignment
2.3. SNP Analysis
2.4. Data Availability
3. Results
3.1. Host Immune Response to SARS-CoV-2 Infection with Gene Editing
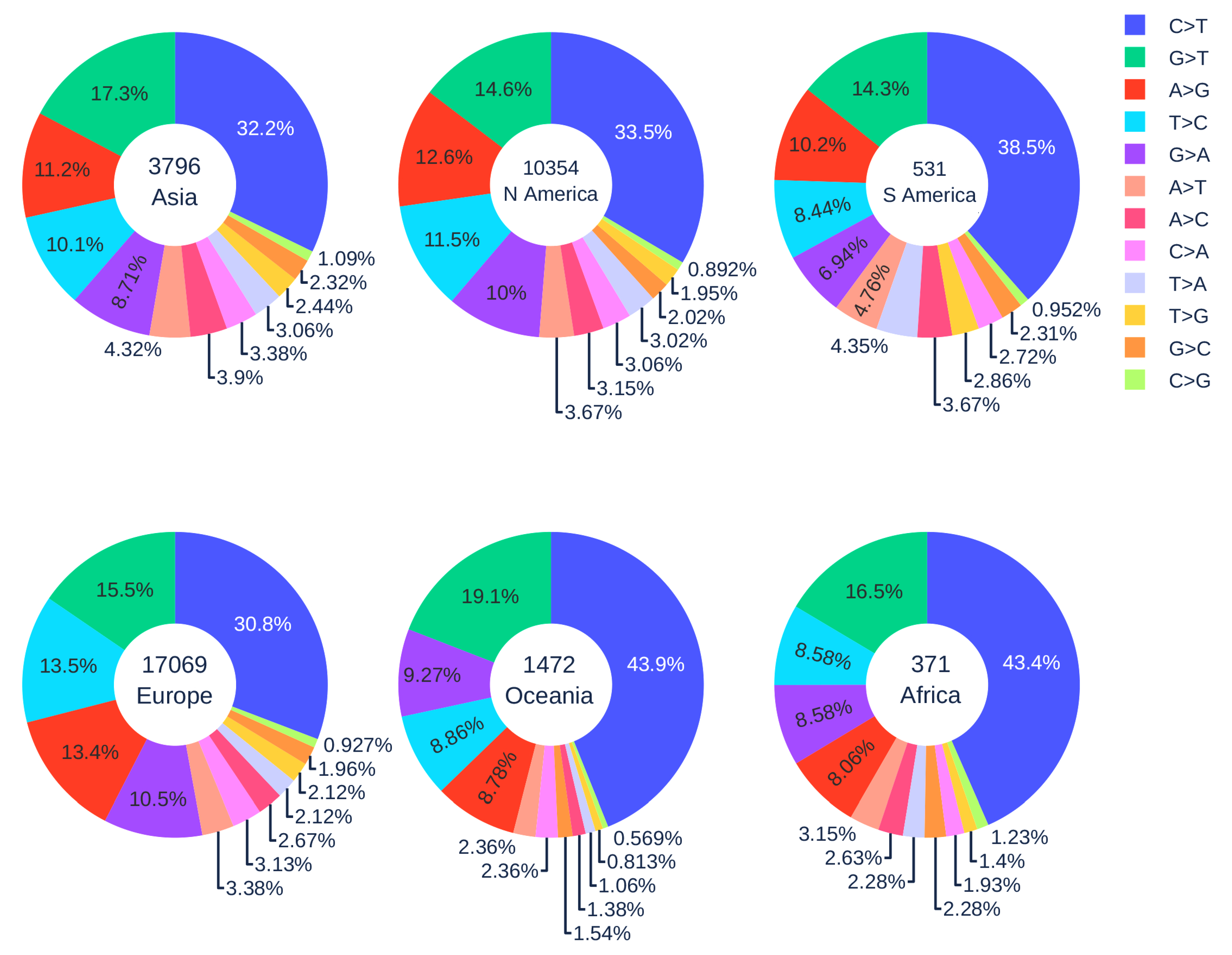
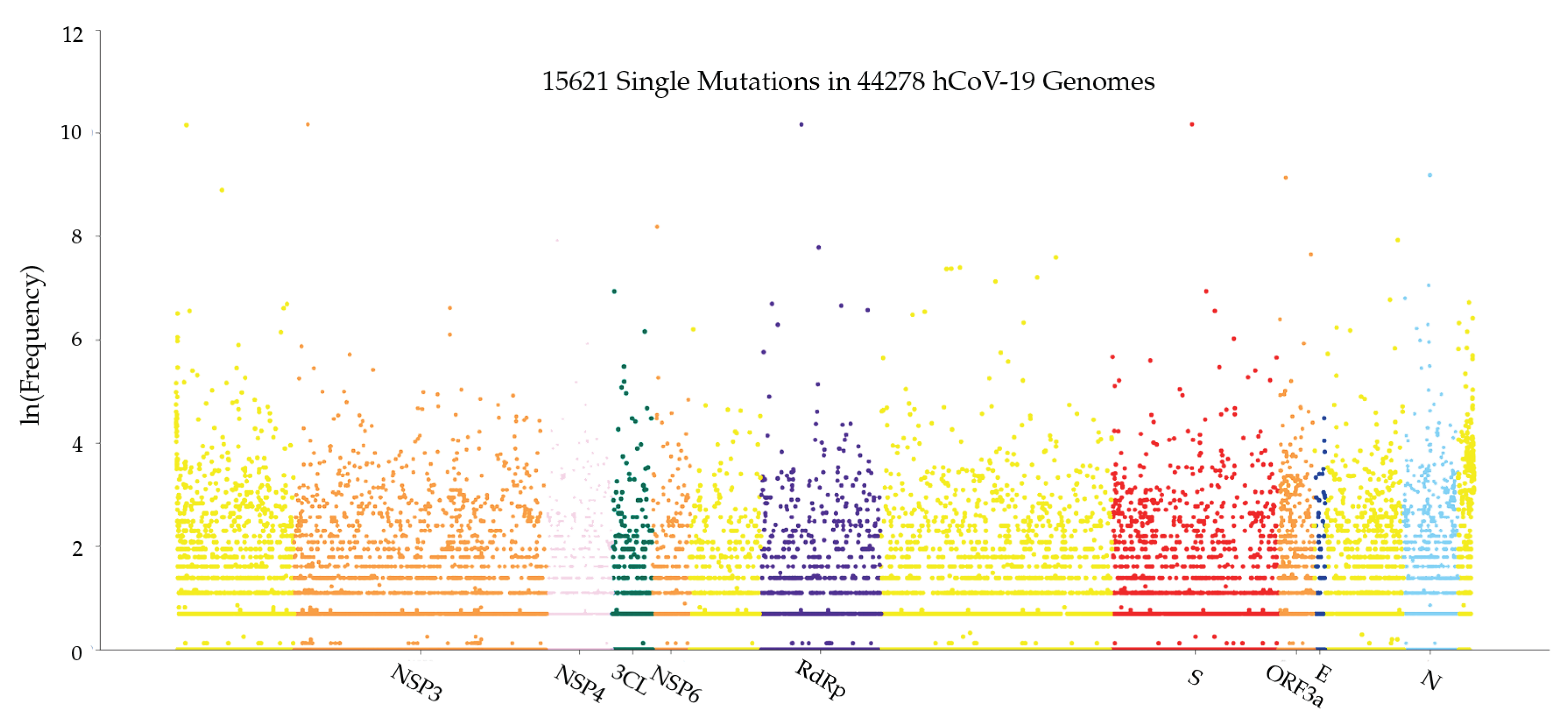
3.1.1. Global Analysis
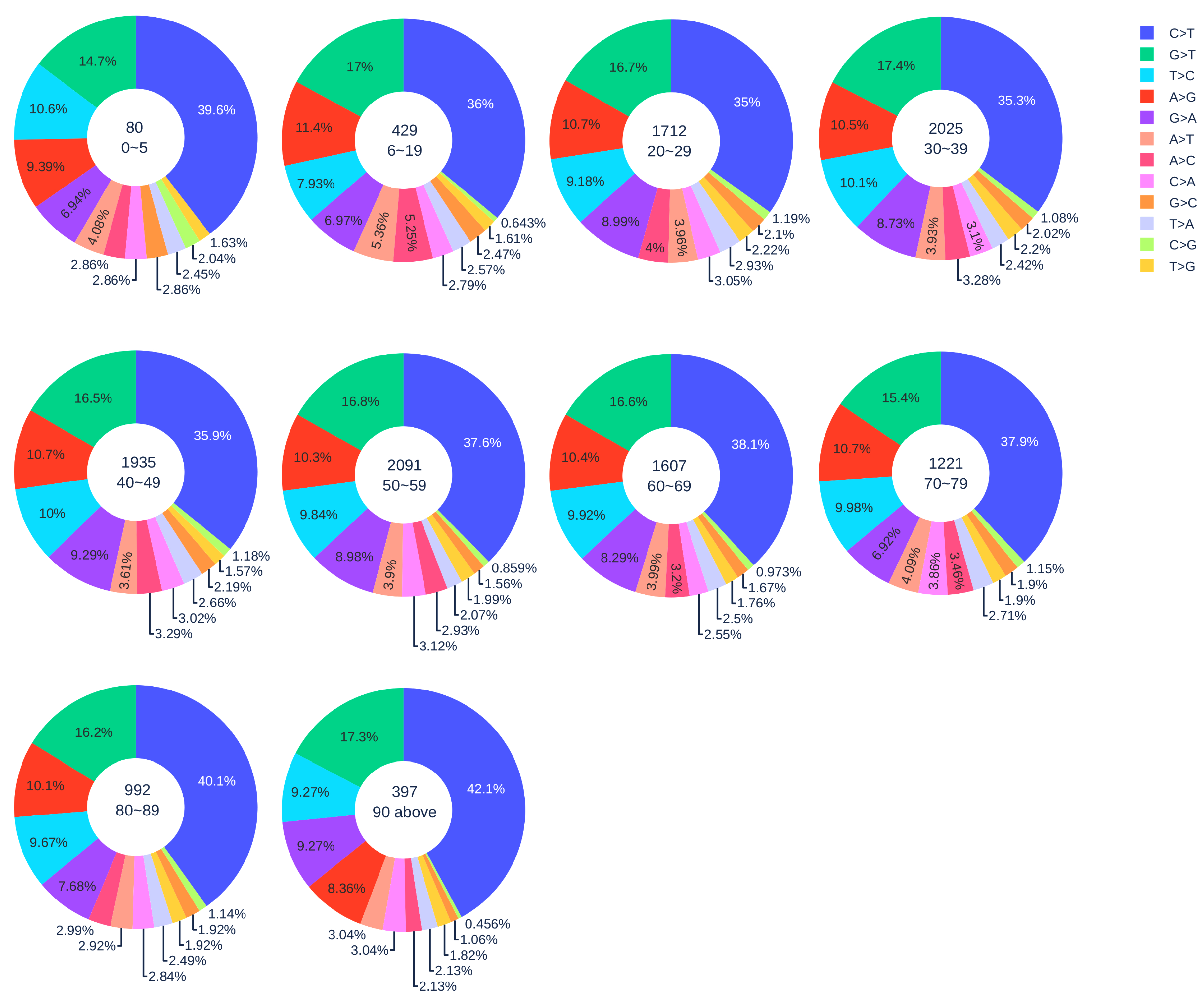
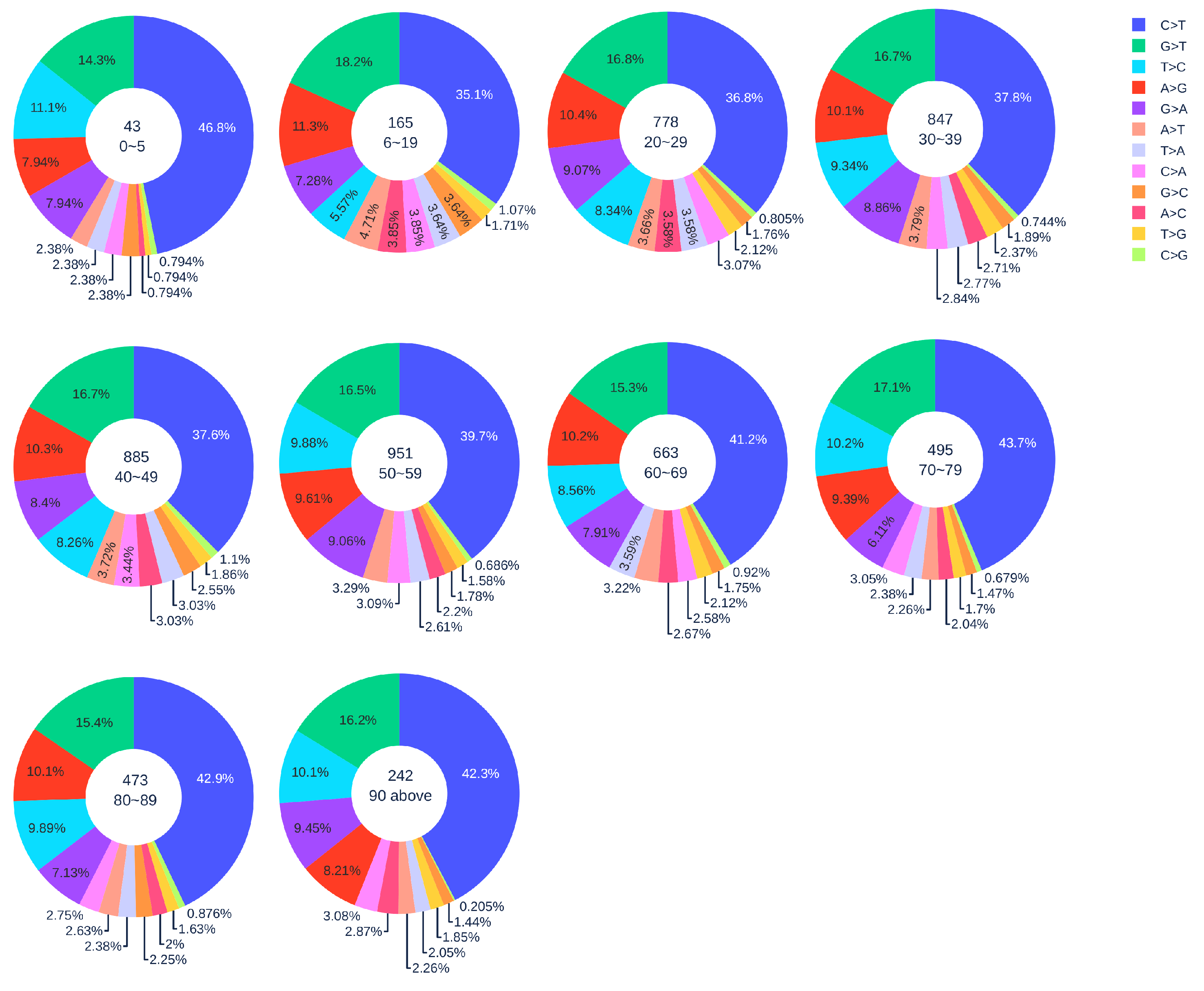
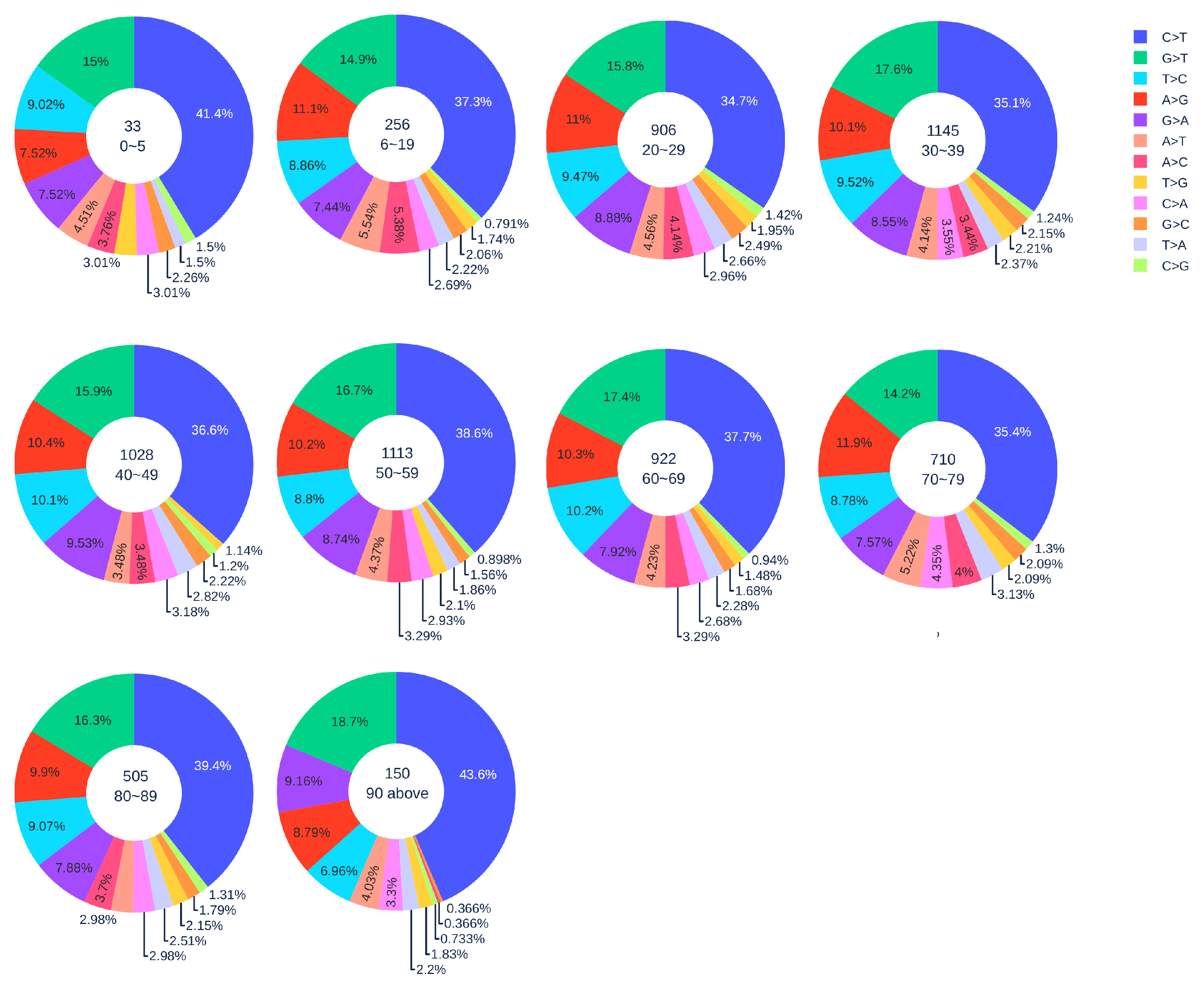
3.1.2. Age Analysis
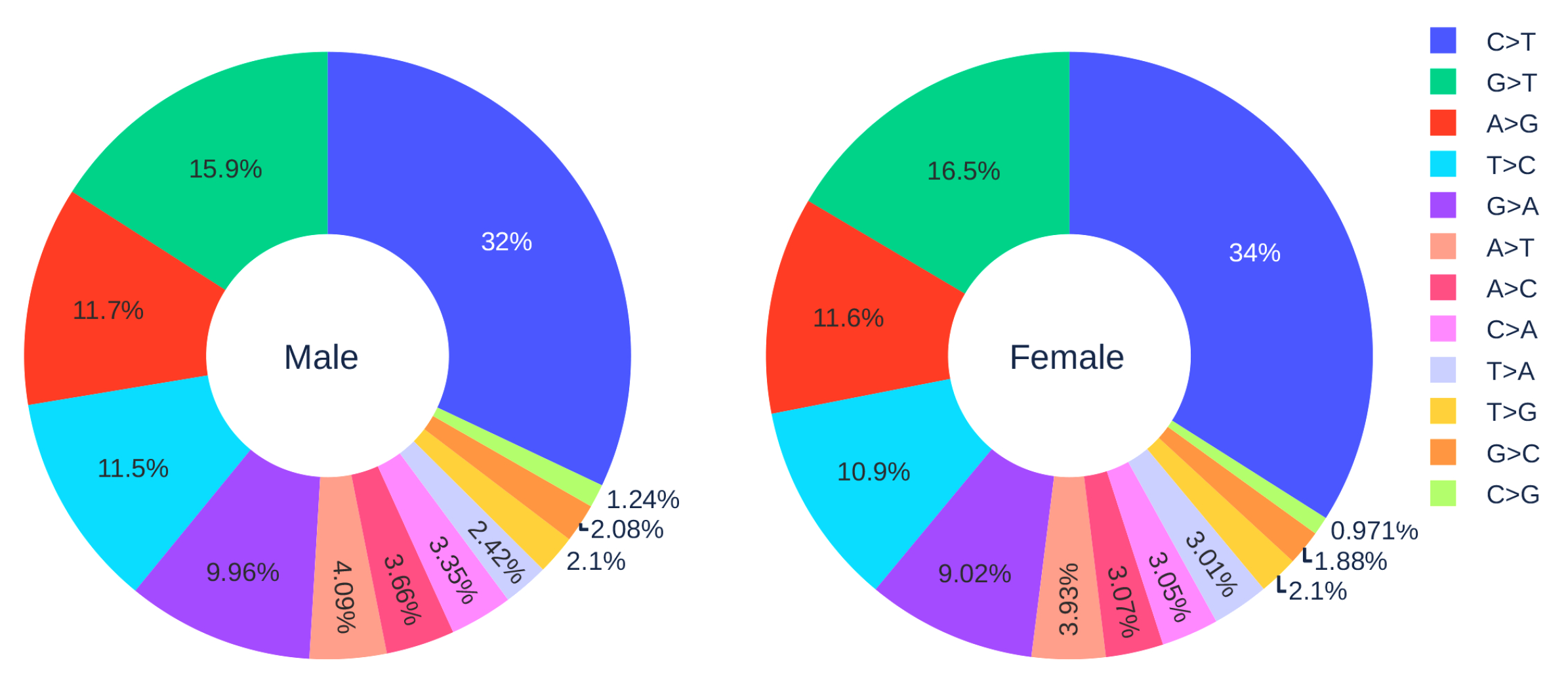
3.1.3. Gender Analysis
3.1.4. Geographic Analysis
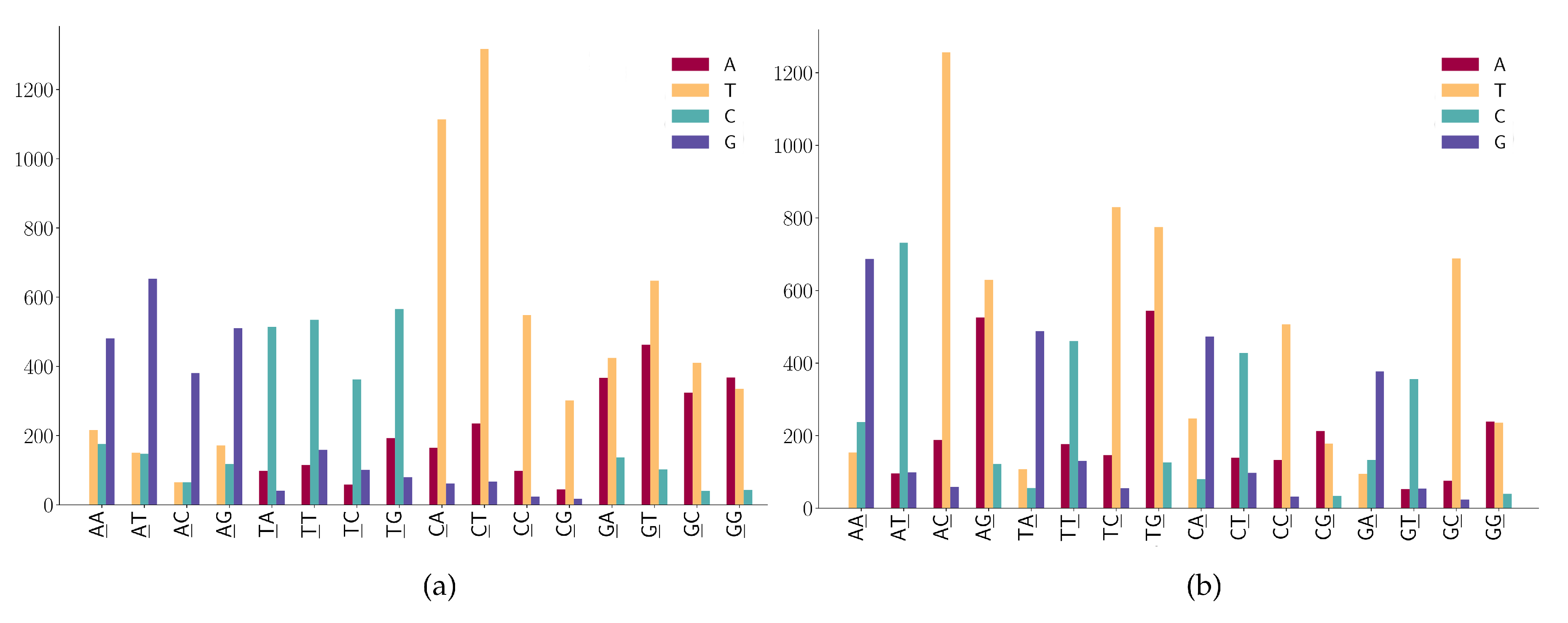
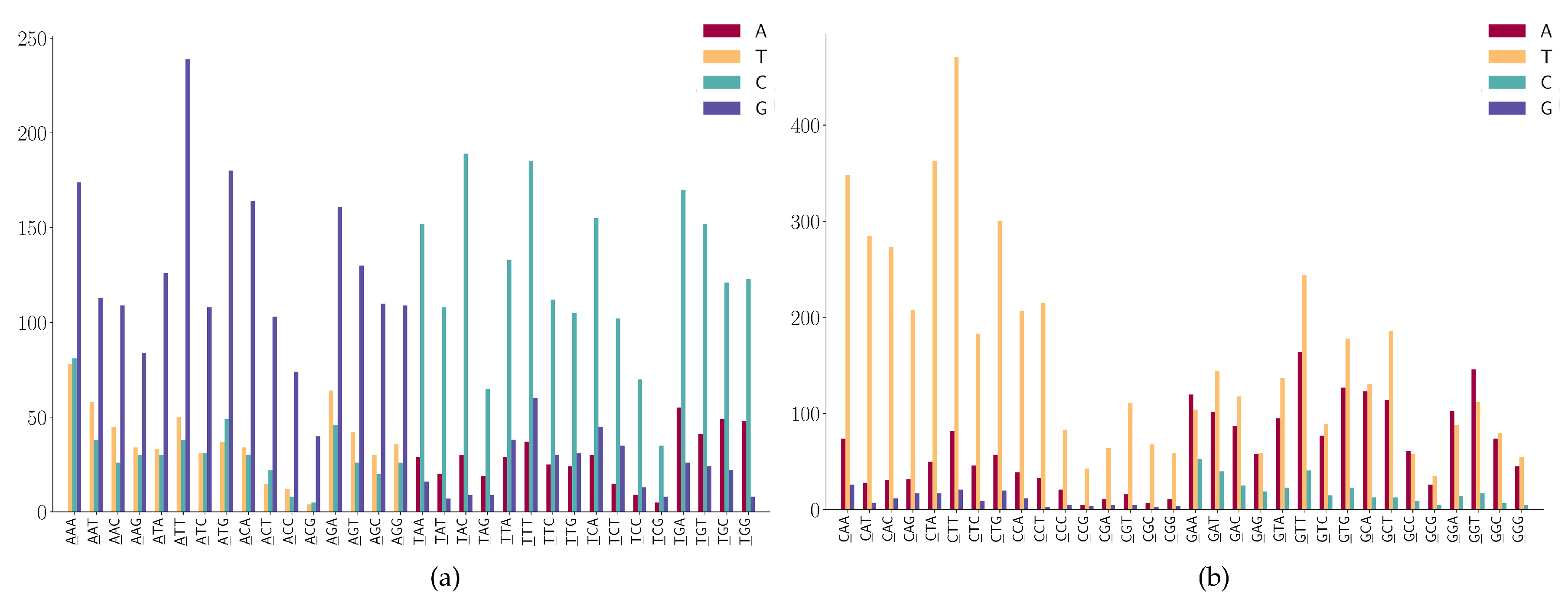
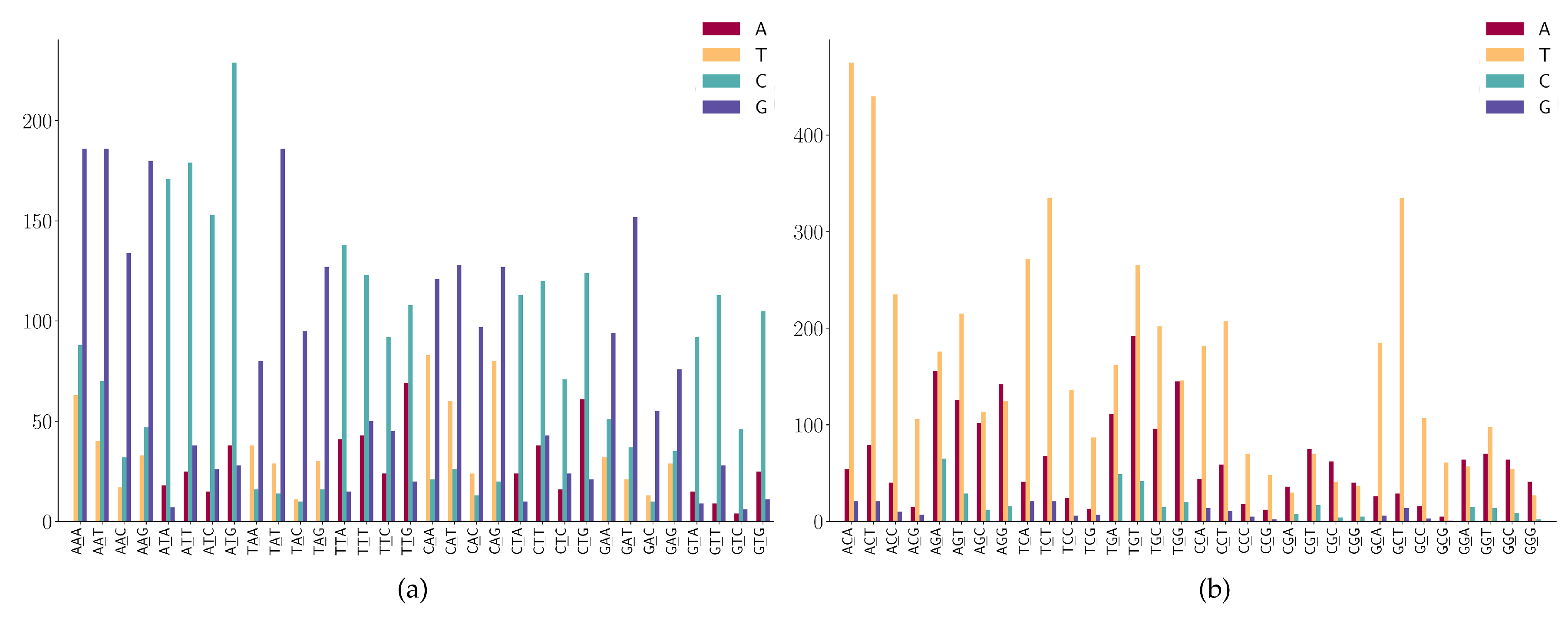
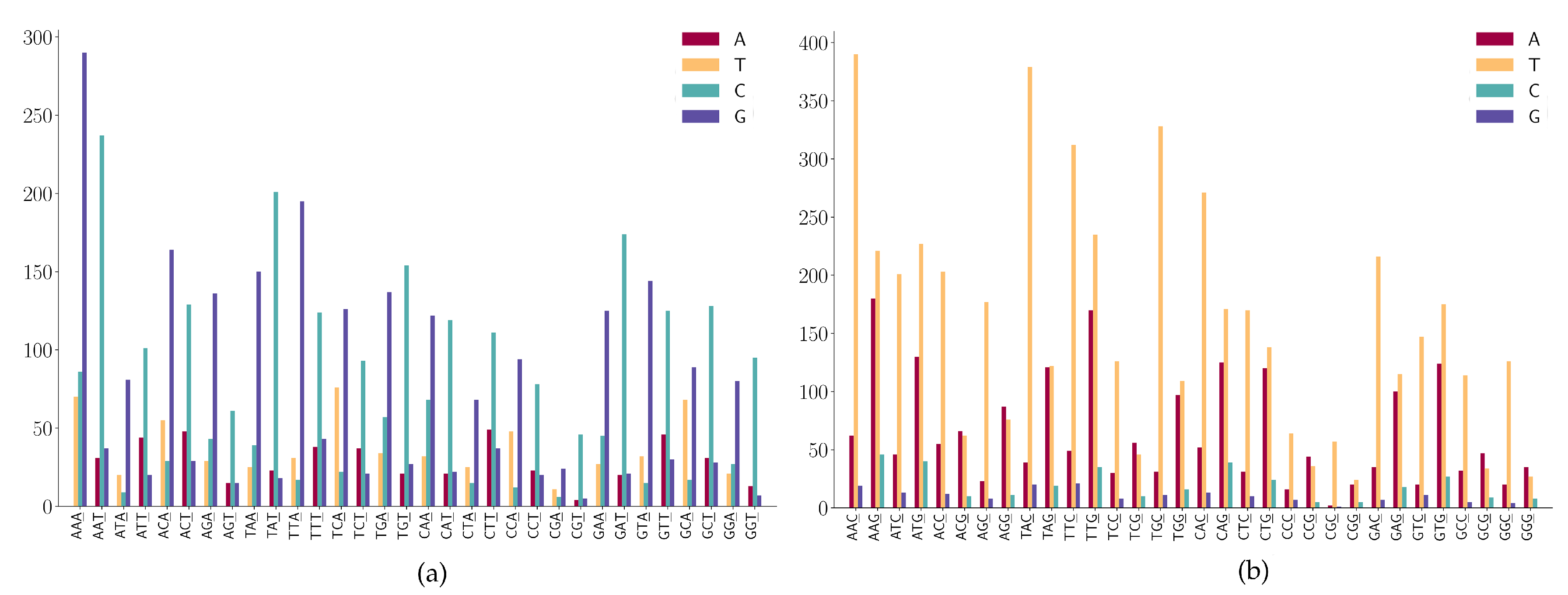
3.2. The SNP Preferences on Sequence Contexts
- (1)
- ANN has high A > G mutations;
- (2)
- TNN has a high frequency in T > C mutations.
- (1)
- CNN has a high frequency in C > T mutations;
- (2)
- GGA and GGT has a high frequency in G > A mutations;
- (3)
- GAA has a relatively high frequency in G > A mutations;
- (4)
- GGW (where W is A or T) has relatively high frequency in G > T mutations.
- (1)
- NAN has a high frequency in A > G mutations;
- (2)
- NTN has a high frequency in T > C mutations;
- (3)
- The A > C mutation also has a larger proportion in AAW (where W is A or T).
- (1)
- WGN (where W is A or T) has a G > T dominated mutation except for AGG;
- (2)
- SGN (where S is G or C) has G > A dominated mutations;
- (3)
- AGG has high G > A mutations;
- (4)
- Characteristic combinations SCG (where S is G or C) are stable and only a few C > G mutations are detected;
- (5)
- Characteristic combinations GGS (where S is G or C) are stable, only a few G > C mutations are detected.
- (1)
- A > G mutation has a high frequency in NNA;
- (2)
- T > C mutation has a high frequency in NNT;
- (3)
- T > C mutation is dominated in NGT and only a few T > A and T > G are found in the sequence context of NGT.
- (1)
- NNC has a high frequency in C > T mutations;
- (2)
- G > T mutation has a high frequency in NNG;
- (3)
- G > A is also highly expressed in the sequence context of NNG;
- (4)
- Characteristic combinations CGC are stable and the mutations on these patterns are most likely to be C > T transitions.
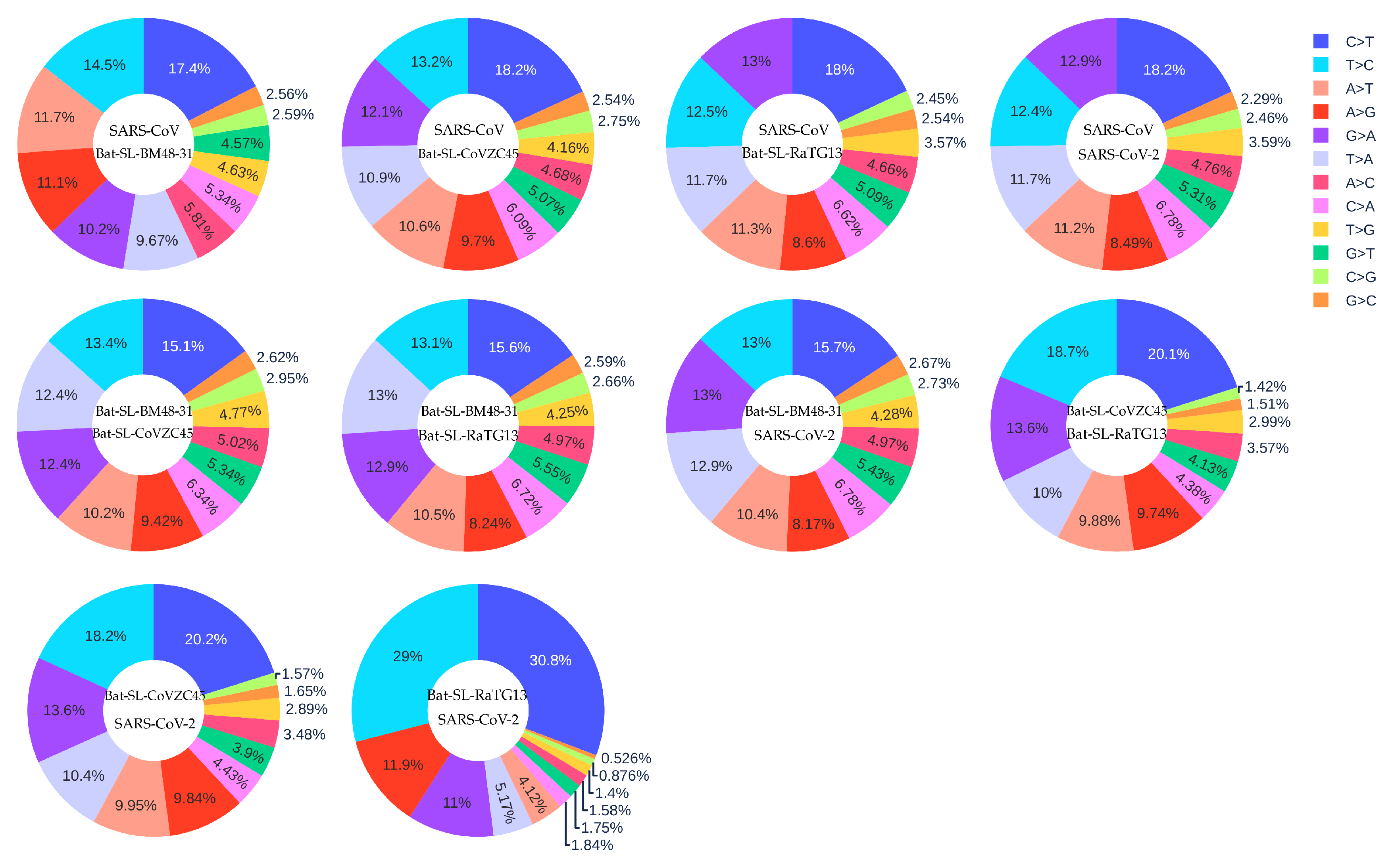
3.3. Coronavirus Evolution
4. Discussion
4.1. Comparison of Unique Mutations and Non-Unique SARS-CoV-2 Mutations
4.2. Comparison of Unique Mutations and Non-Unique MERS-CoV-2 Mutations
4.3. Gene- and Protein-Specific Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| COVID-19 | Coronavirus disease 2019 |
| APOBEC | Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like |
| ADAR | Adenosine deaminases acting on RNA |
| AIDs | Activation-induced cytidine deaminases |
References
- WHO. Coronavirus Disease 2019 (COVID-19) Situation Report—193; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Chakraborti, S.; Dimitrov, A.S.; Gramatikoff, K.; Dimitrov, D.S. The SARS-CoV S glycoprotein: Expression and functional characterization. Biochem. Biophys. Res. Commun. 2003, 312, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Glowacka, I.; Bertram, S.; Müller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- McBride, R.; Van Zyl, M.; Fielding, B.C. The coronavirus nucleocapsid is a multifunctional protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef] [Green Version]
- Yue, P.; Li, Z.; Moult, J. Loss of protein structure stability as a major causative factor in monogenic disease. J. Mol. Biol. 2005, 353, 459–473. [Google Scholar] [CrossRef]
- Stefl, S.; Nishi, H.; Petukh, M.; Panchenko, A.R.; Alexov, E. Molecular mechanisms of disease-causing missense mutations. J. Mol. Biol. 2013, 425, 3919–3936. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Hozumi, Y.; Yin, C.; Wei, G.-W. Mutations on COVID-19 diagnostic targets. Genomics. 2020, in press. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking changes in SARS-CoV-2 Spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Hanage, W.P.; Rasmussen, A.L. Making sense of mutation: What D614G means for the COVID-19 pandemic remains unclear. Cell 2020, 182, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.-W. Mutations strengthened SARS-CoV-2 infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, Y.M.; Flamholz, A.; Phillips, R.; Milo, R. Science Forum: SARS-CoV-2 (COVID-19) by the numbers. Elife 2020, 9, e57309. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, J.; Zhu, A.; Zhao, J.; Zhao, J. Current understanding of middle east respiratory syndrome coronavirus infection in human and animal models. J. Thorac. Dis. 2018, 10 (Suppl. 19), S2260. [Google Scholar] [CrossRef] [PubMed]
- Sevajol, M.; Subissi, L.; Decroly, E.; Canard, B.; Imbert, I. Insights into RNA synthesis, capping, and proofreading mechanisms of SARS-coronavirus. Virus Res. 2014, 194, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Ferron, F.; Subissi, L.; De Morais, A.T.S.; Le, N.T.T.; Sevajol, M.; Gluais, L.; Decroly, E.; Vonrhein, C.; Bricogne, G.; Canard, B.; et al. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus rna. Proc. Natl. Acad. Sci. USA 2018, 115, E162–E171. [Google Scholar] [CrossRef] [Green Version]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [Green Version]
- Yin, C. Genotyping coronavirus SARS-CoV-2: Methods and implications. Genomics 2020, 112, 3588–3596. [Google Scholar] [CrossRef]
- Phan, T. Genetic diversity and evolution of SARS-CoV-2. Infect. Genet. Evol. 2020, 81, 104260. [Google Scholar] [CrossRef]
- Khan, K.A.; Cheung, P. Presence of mismatches between diagnostic PCR assays and coronavirus sars-cov-2 genome. R. Soc. Open Sci. 2020, 7, 200636. [Google Scholar] [CrossRef]
- Wang, R.; Hozumi, Y.; Yin, C.; Wei, G.-W. Decoding SARS-CoV-2 transmission, evolution, and ramification on COVID-19 diagnosis, vaccine, and medicine. J. Chem. Inf. Model. 2020. [Google Scholar] [CrossRef]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef]
- Di Giorgio, S.; Martignano, F.; Torcia, M.G.; Mattiuz, G.; Conticello, S.G. Evidence for RNA editing in the transcriptome of 2019 novel coronavirus. Sci. Adv. 2020, 6, eabb5813. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. Rampant C > U hypermutation in the genomes of SARS-CoV-2 and other coronaviruses: Causes and consequences for their short-and long-term evolutionary trajectories. Msphere 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Matyášek, R.; Kovařík, A. Mutation patterns of human SARS-CoV-2 and Bat RaTG13 coronavirus genomes are strongly biased towards C > U transitions, indicating rapid evolution in their hosts. Genes 2020, 11, 761. [Google Scholar] [CrossRef]
- Zheng, Y.-H.; Irwin, D.; Kurosu, T.; Tokunaga, K.; Sata, T.; Peterlin, B.M. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J. Virol. 2004, 78, 6073–6076. [Google Scholar] [CrossRef] [Green Version]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, H.C.; Bennett, R.P.; Kizilyer, A.; McDougall, W.M.; Prohaska, K.M. Functions and regulation of the APOBEC family of proteins. In Seminars in Cell & Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2012; Volume 23, pp. 258–268. [Google Scholar]
- Liu, M.-C.; Liao, W.-Y.; Buckley, K.M.; Yang, S.Y.; Rast, J.P.; Fugmann, S.D. AID/APOBEC-like cytidine deaminases are ancient innate immune mediators in invertebrates. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Samuel, C.E. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 2011, 411, 180–193. [Google Scholar] [CrossRef]
- Gonzales-van Horn, S.R.; Sarnow, P. Making the mark: The role of adenosine modifications in the life cycle of RNA viruses. Cell Host Microbe 2017, 21, 661–669. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479, 131–145. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Li, W.; Xie, J.; Hou, Y.; You, C. Cytokine storm induced by SARS-CoV-2. Clin. Chim. Acta 2020, 509, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; McCauley, J. Gisaid: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, F.; Higgins, D.G. Clustal omega. Curr. Protoc. Bioinform. 2014, 48, 3–13. [Google Scholar] [CrossRef]
- Söding, J. Protein homology detection by hmm–hmm comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef] [Green Version]
- Blackshields, G.; Sievers, F.; Shi, W.; Wilm, A.; Higgins, D.G. Sequence embedding for fast construction of guide trees for multiple sequence alignment. Algorithms Mol. Biol. 2010, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Hewagama, A.; Patel, D.; Yarlagadda, S.; Strickland, F.M.; Richardson, B.C. Stronger inflammatory/cytotoxic T-cell response in women identified by microarray analysis. Genes Immun. 2009, 10, 509–516. [Google Scholar] [CrossRef]
- Klein, S.L. Sex influences immune responses to viruses, and efficacy of prophylaxis and treatments for viral diseases. Bioessays 2012, 34, 1050–1059. [Google Scholar] [CrossRef] [Green Version]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Lee, N.; Hui, D.; Wu, A.; Chan, P.; Cameron, P.; Joynt, G.M.; Ahuja, A.; Yung, M.Y.; Leung, C.B.; To, K.F.; et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003, 348, 1986–1994. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Gloza-Rausch, F.; Glende, J.; Corman, V.M.; Muth, D.; Goettsche, M.; Seebens, A.; Niedrig, M.; Pfefferle, S.; Yordanov, S.; et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 2010, 84, 11336–11349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, D.; Zhu, C.; Ai, L.; He, T.; Wang, Y.; Ye, F.; Yang, L.; Ding, C.; Zhu, X.; Lv, R.; et al. Genomic characterization and infectivity of a novel SARS-like coronavirus in Chinese bats. Emerg. Microbes Infect. 2018, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Wang, A.; Liu, M.; Wang, Q.; Chen, J.; Xia, S.; Ling, Y.; Zhang, Y.; Xun, J.; Lu, L.; et al. Neutralizing antibody responses to SARS-CoV-2 in a COVID-19 recovered patient cohort and their implications. medRxiv 2020. [Google Scholar] [CrossRef]
- Bai, Y.; Yao, L.; Wei, T.; Tian, F.; Jin, D.-Y.; Chen, L.; Wang, M. Presumed asymptomatic carrier transmission of COVID-19. JAMA 2020, 323, 1406–1407. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
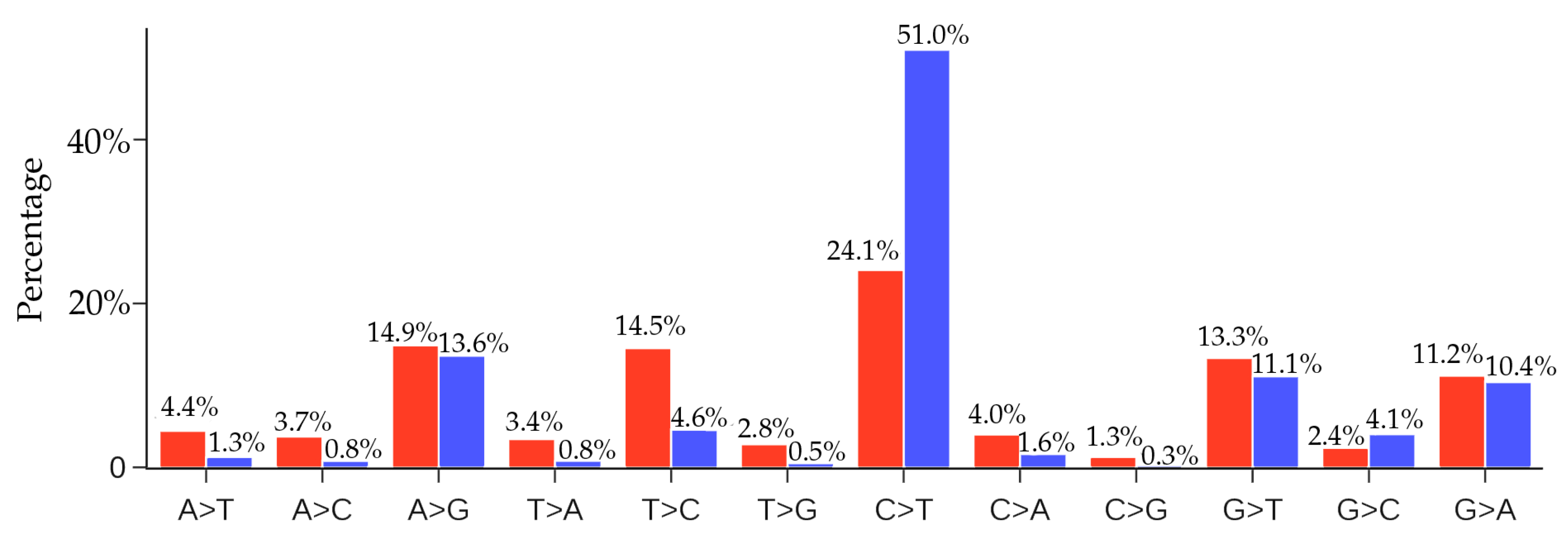
| SNP Type | Mutation Type | Ratio | SNP Type | Mutation Type | Ratio |
|---|---|---|---|---|---|
| A > T | Transversion | 4.44% | C > T | Transition | 24.06% |
| A > C | Transversion | 3.75% | C > A | Transversion | 4.00% |
| A > G | Transition | 14.87% | C > G | Transversion | 1.25% |
| T > A | Transversion | 3.43% | G > T | Transversion | 13.33% |
| T > C | Transition | 14.53% | G > C | Transversion | 2.36% |
| T > G | Transversion | 2.80% | G > A | Transition | 11.17% |
| Country | Total Counts | Age Counts | Gender Counts |
|---|---|---|---|
| United Kingdom | 10,740 | 2159 | 2134 |
| United States | 8729 | 1888 | 2095 |
| Australia | 1329 | 776 | 750 |
| India | 1088 | 1068 | 1071 |
| World | 33,693 | 12,513 | 12,181 |
| Country | 0–19 | 20–29 | 30–39 | 40–49 | 50–59 | 60–69 | 70–79 | 80 above |
|---|---|---|---|---|---|---|---|---|
| United Kingdom | 44.4% | 48.4% | 48.7% | 49.8% | 46.9% | 46.6% | 48.0% | 41.7% |
| United States | 51.0% | 45.4% | 44.1% | 40.0% | 41.6% | 40.7% | 47.6% | 45.6% |
| Australia | 35.8% | 43.1% | 44.1% | 41.6% | 45.5% | 41.7% | 45.6% | 42.0% |
| India | 39.0% | 38.2% | 38.2% | 35.2% | 42.4% | 40.9% | 46.7% | 55.0% |
| From | To | C > T Ratio | T > C Ratio | C > T/T > C Ratio |
|---|---|---|---|---|
| SARS-CoV-2 reference genome | 33693 SARS-CoV-2 genomes | 24.06% | 14.53% | 1.66 |
| SARS-CoV | Bat-SL-BM48-31 | 17.40% | 14.50% | 1.20 |
| SARS-CoV | Bat-SL-CoVZC45 | 18.20% | 13.20% | 1.37 |
| SARS-CoV | Bat-SL-RaTG13 | 18.00% | 12.50% | 1.50 |
| SARS-CoV | SARS-CoV-2 | 18.20% | 12.40% | 1.47 |
| Bat-SL-BM48-31 | Bat-SL-CoVZC45 | 15.10% | 13.40% | 1.13 |
| Bat-SL-BM48-31 | Bat-SL-RaTG13 | 15.60% | 13.10% | 1.19 |
| Bat-SL-BM48-31 | SARS-CoV-2 | 15.70% | 13.00% | 1.21 |
| Bat-SL-CoVZC45 | Bat-SL-RaTG13 | 20.10% | 18.70% | 1.07 |
| Bat-SL-CoVZC45 | SARS-CoV-2 | 20.20% | 18.20% | 1.11 |
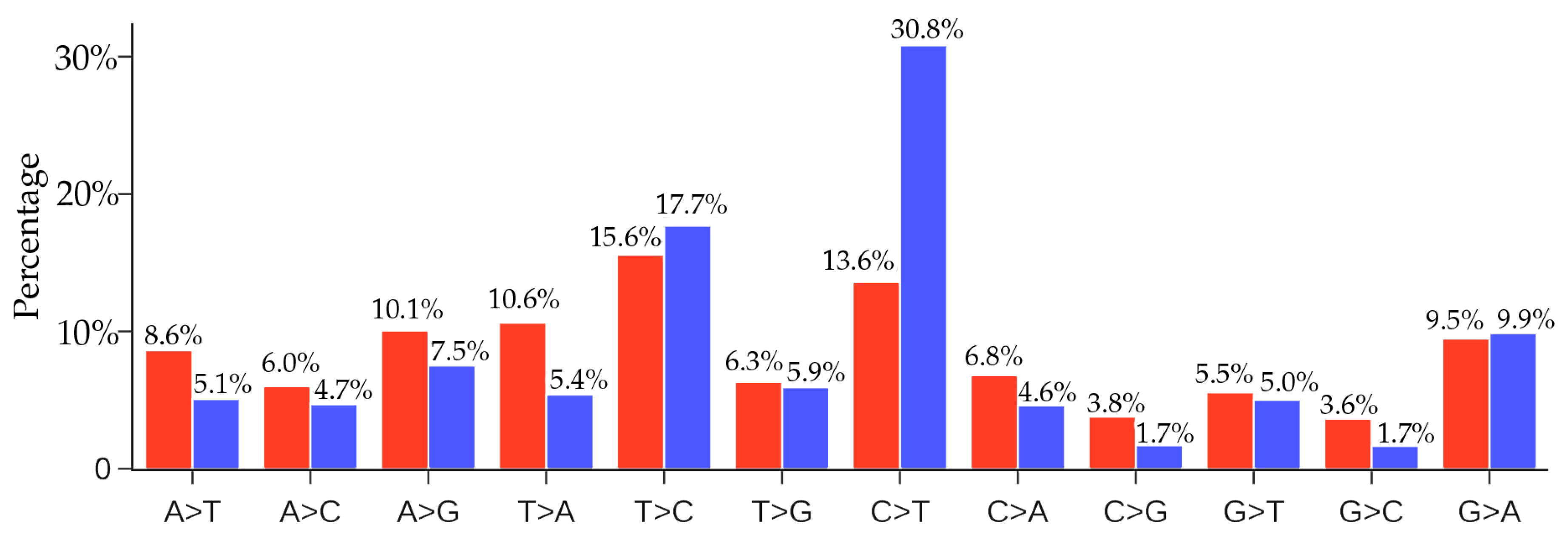
| Bat-SL-RaTG13 | SARS-CoV-2 | 30.80% | 29.00% | 1.06 |
| Gene Type | Gene Site | Gene Length | C > T Ratio | Corrected C > T Ratio |
|---|---|---|---|---|
| NSP1 | 266:805 | 540 | 29.0% | 1.35 |
| NSP2 | 806:2719 | 1914 | 26.1% | 1.41 |
| NSP3 | 2720:8554 | 5835 | 25.4% | 1.52 |
| NSP4 | 8555:10054 | 1500 | 30.7% | 1.73 |
| NSP5(3CL) | 10055:10972 | 918 | 32.7% | 1.84 |
| NSP6 | 10973:11842 | 870 | 23.7% | 1.43 |
| NSP7 | 11843:12091 | 249 | 29.0% | 1.47 |
| NSP8 | 12092:12685 | 594 | 28.4% | 1.58 |
| NSP9 | 12686:13024 | 339 | 34.4% | 1.82 |
| NSP10 | 13025:13441 | 417 | 30.4% | 1.51 |
| NSP11 | 13442:13480 | 39 | 33.3% | 1.86 |
| RNA-dependent-polymerase | 13442:16236 | 2796 | 25.4% | 1.42 |
| Helicase | 16237:18039 | 1803 | 26.5% | 1.42 |
| 3’-to-5’ exonuclease | 18040:19620 | 1581 | 27.1% | 1.48 |
| endoRNAse | 19621:20658 | 1038 | 19.8% | 1.37 |
| 2’-O-ribose methyltransferase | 20659:21552 | 894 | 24.3% | 1.52 |
| Spike protein | 21563:25384 | 3819 | 21.3% | 1.13 |
| ORF3a protein | 25393:26220 | 825 | 22.8% | 1.08 |
| Envelope protein | 26245:26472 | 225 | 22.4% | 1.14 |
| Membrane glycoprotein | 26523:27191 | 666 | 26.4% | 1.21 |
| ORF6 protein | 27202:27387 | 183 | 16.6% | 1.19 |
| ORF7a protein | 27394:27759 | 363 | 21.9% | 1.04 |
| ORF7b protein | 27756:27887 | 129 | 8.3% | 0.67 |
| ORF8 protein | 27894:28259 | 363 | 18.1% | 1.03 |
| Nucleocapsid protein | 28274:29533 | 1257 | 24.0% | 0.96 |
| ORF10 protein | 29558:29674 | 114 | 28.4% | 1.58 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Hozumi, Y.; Zheng, Y.-H.; Yin, C.; Wei, G.-W. Host Immune Response Driving SARS-CoV-2 Evolution. Viruses 2020, 12, 1095. https://doi.org/10.3390/v12101095
Wang R, Hozumi Y, Zheng Y-H, Yin C, Wei G-W. Host Immune Response Driving SARS-CoV-2 Evolution. Viruses. 2020; 12(10):1095. https://doi.org/10.3390/v12101095
Chicago/Turabian StyleWang, Rui, Yuta Hozumi, Yong-Hui Zheng, Changchuan Yin, and Guo-Wei Wei. 2020. "Host Immune Response Driving SARS-CoV-2 Evolution" Viruses 12, no. 10: 1095. https://doi.org/10.3390/v12101095
APA StyleWang, R., Hozumi, Y., Zheng, Y. -H., Yin, C., & Wei, G. -W. (2020). Host Immune Response Driving SARS-CoV-2 Evolution. Viruses, 12(10), 1095. https://doi.org/10.3390/v12101095