Unmapped RNA Virus Diversity in Termites and Their Symbionts

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Termite Collection, RNA Isolation and Sequencing
2.2. Assembly and Virus Identification
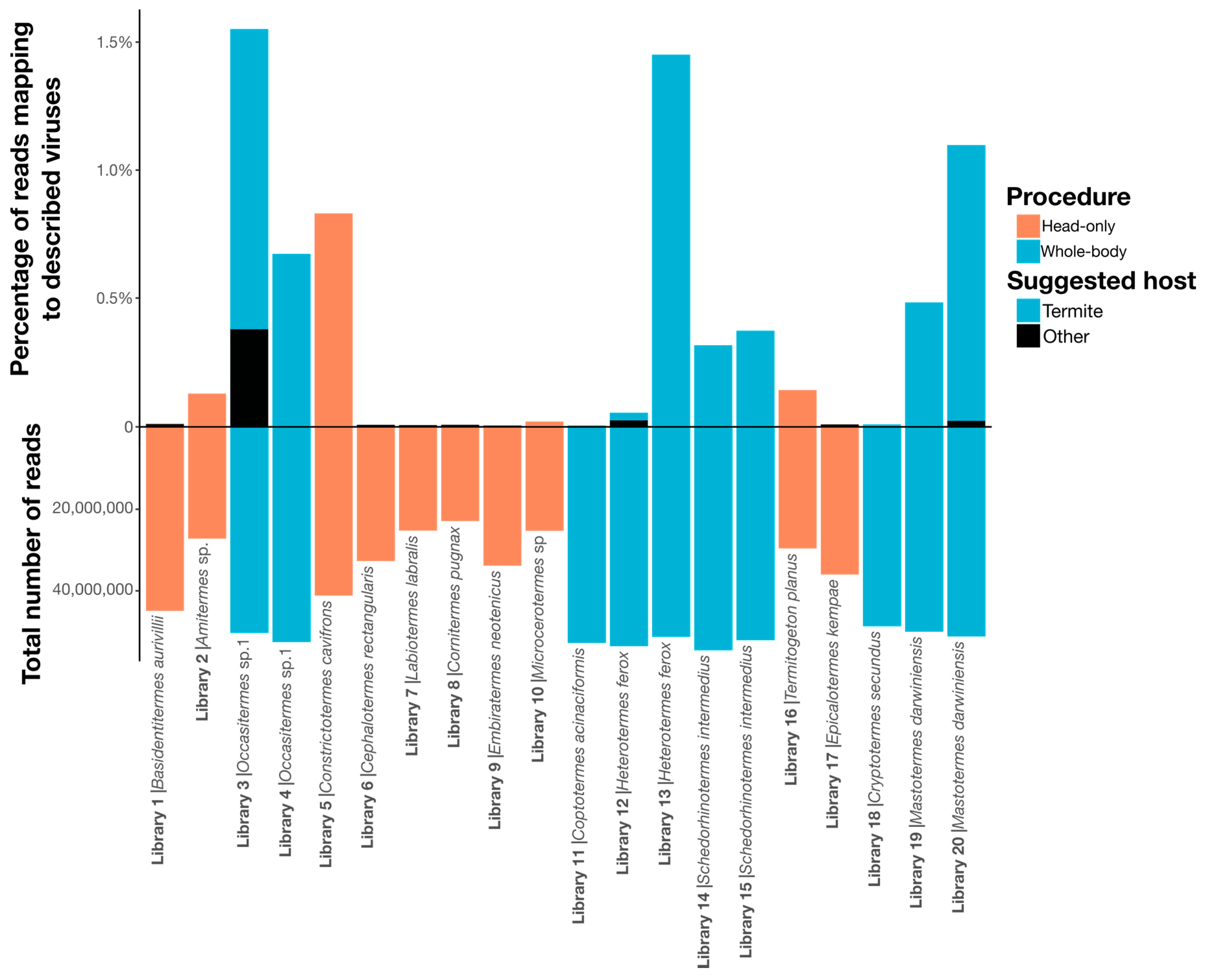
2.3. Virus Abundance Measurements
2.4. Viruses Excluded from the Analysis
2.5. Phylogenetic Analysis
2.6. Statistical Analysis of Abundance and Diversity
2.7. Statistical Analysis of Abundance and Diversity
3. Results
3.1. Identification and Annotation of RNA Viruses
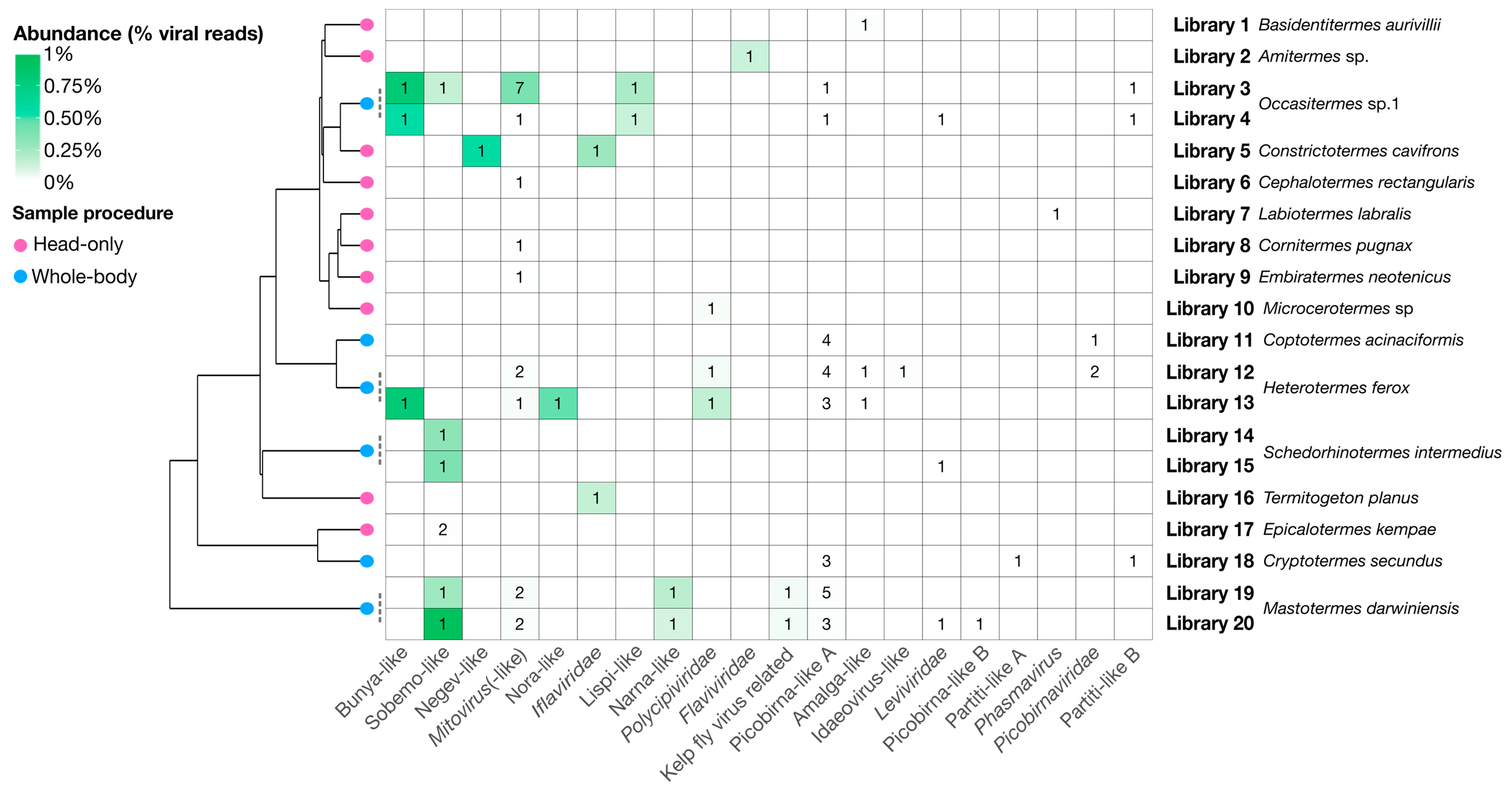
3.2. RNA Virus Diversity in Termites
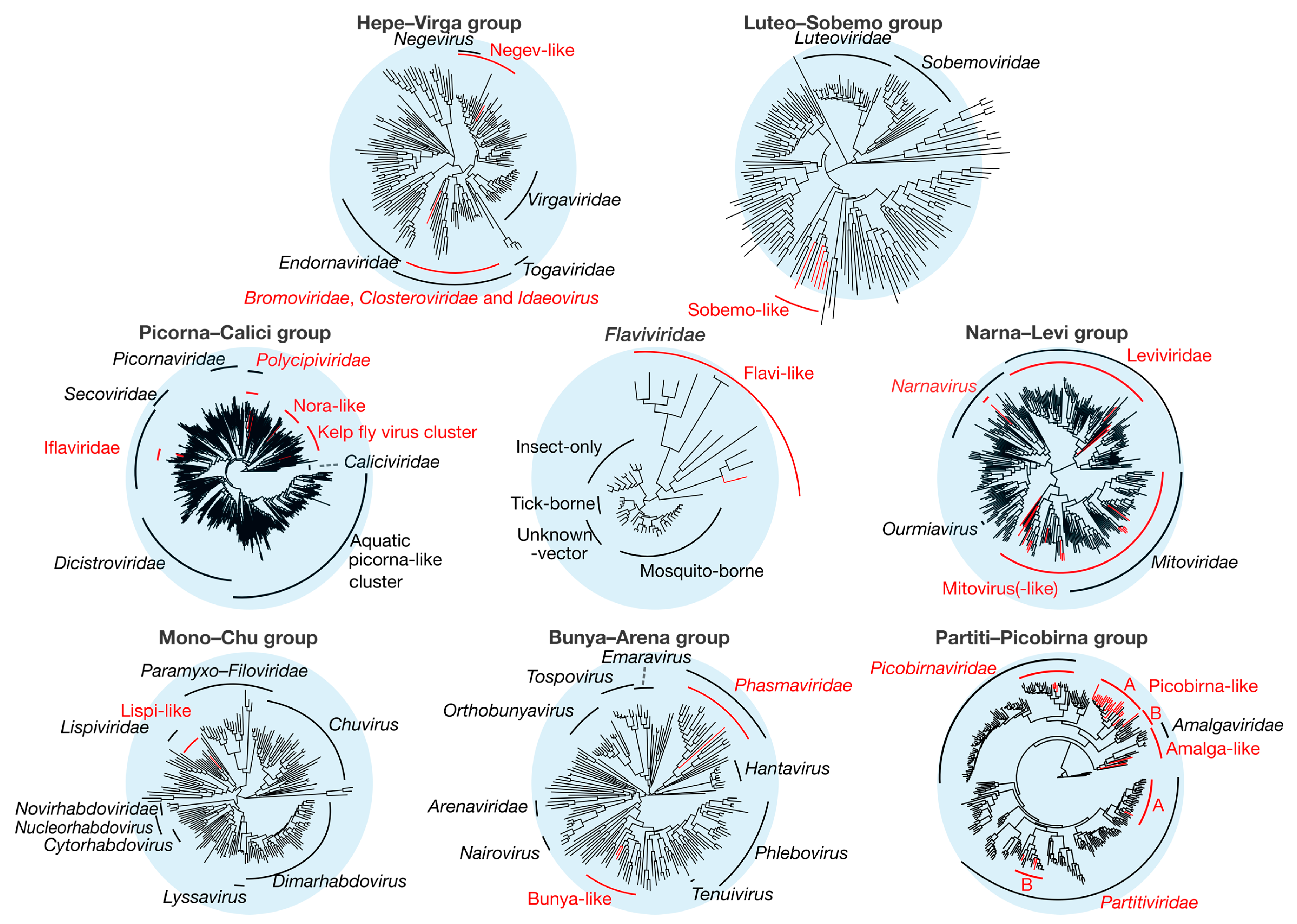
3.3. Positive-Sense ssRNA Viruses
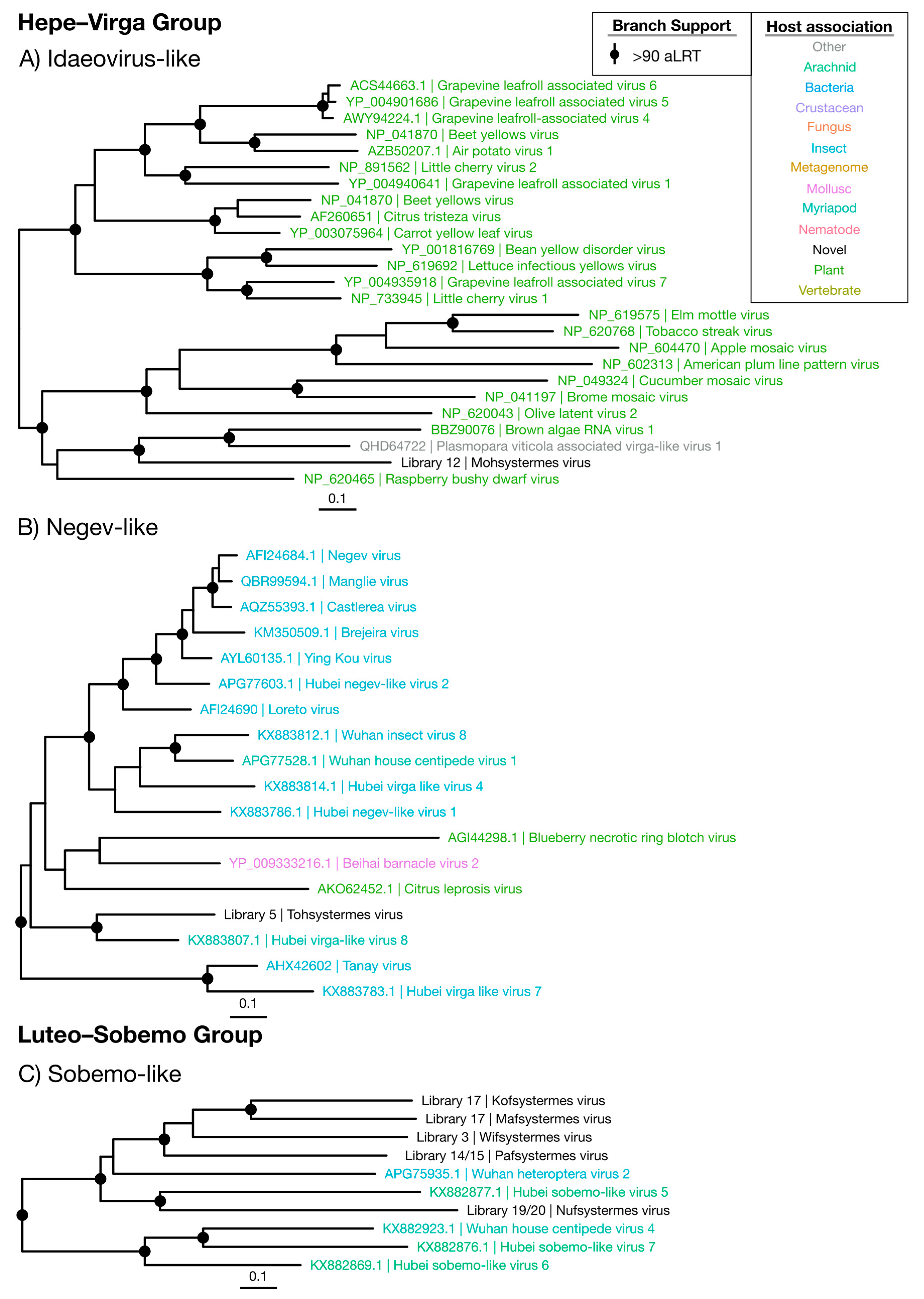
3.3.1. Hepe–Virga Viruses
3.3.2. Luteo–Sobemo Viruses
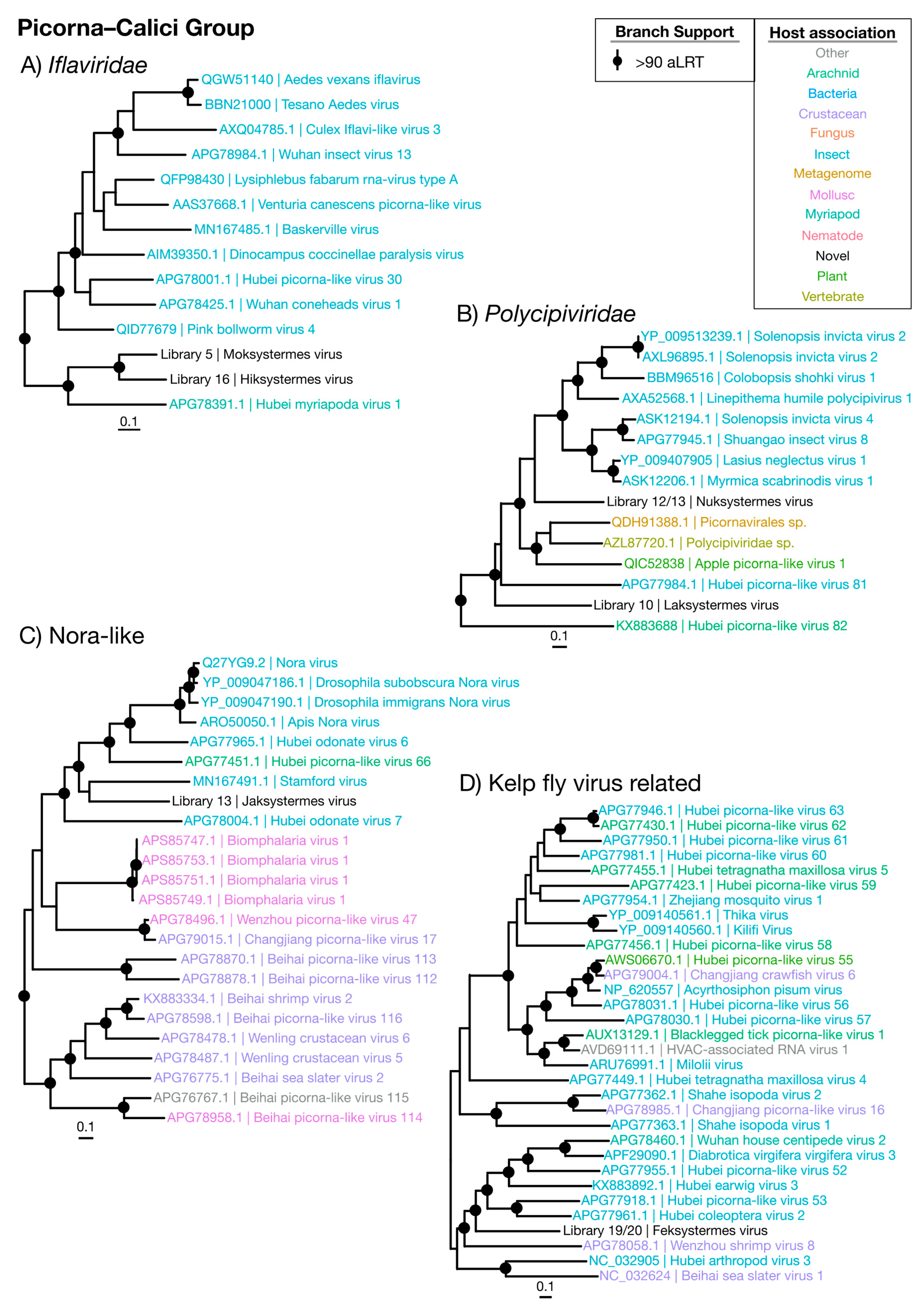
3.3.3. Picorna–Calici Viruses
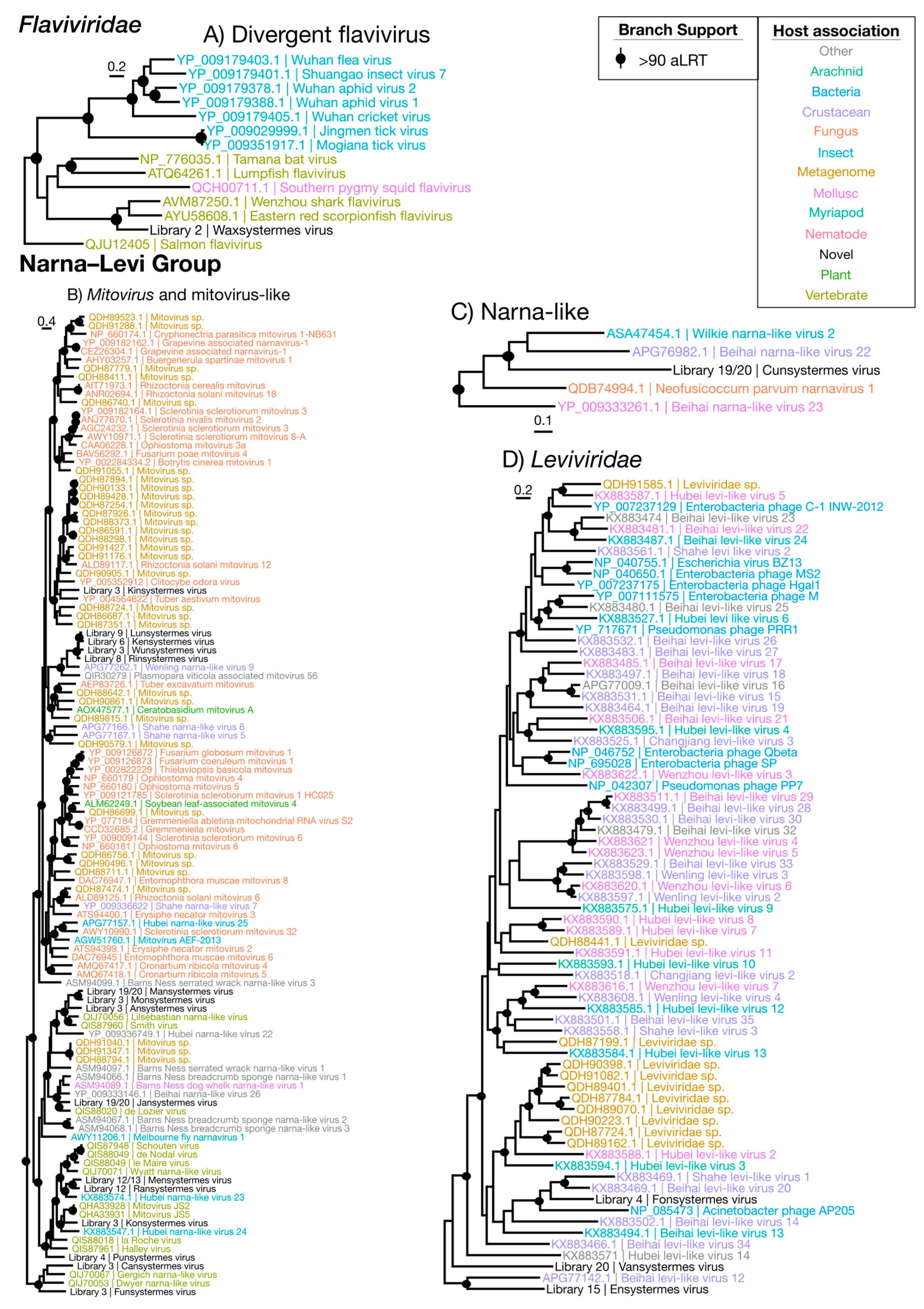
3.3.4. Flaviviridae
3.3.5. Narna–Levi Viruses
3.4. Negative-Sense ssRNA Viruses
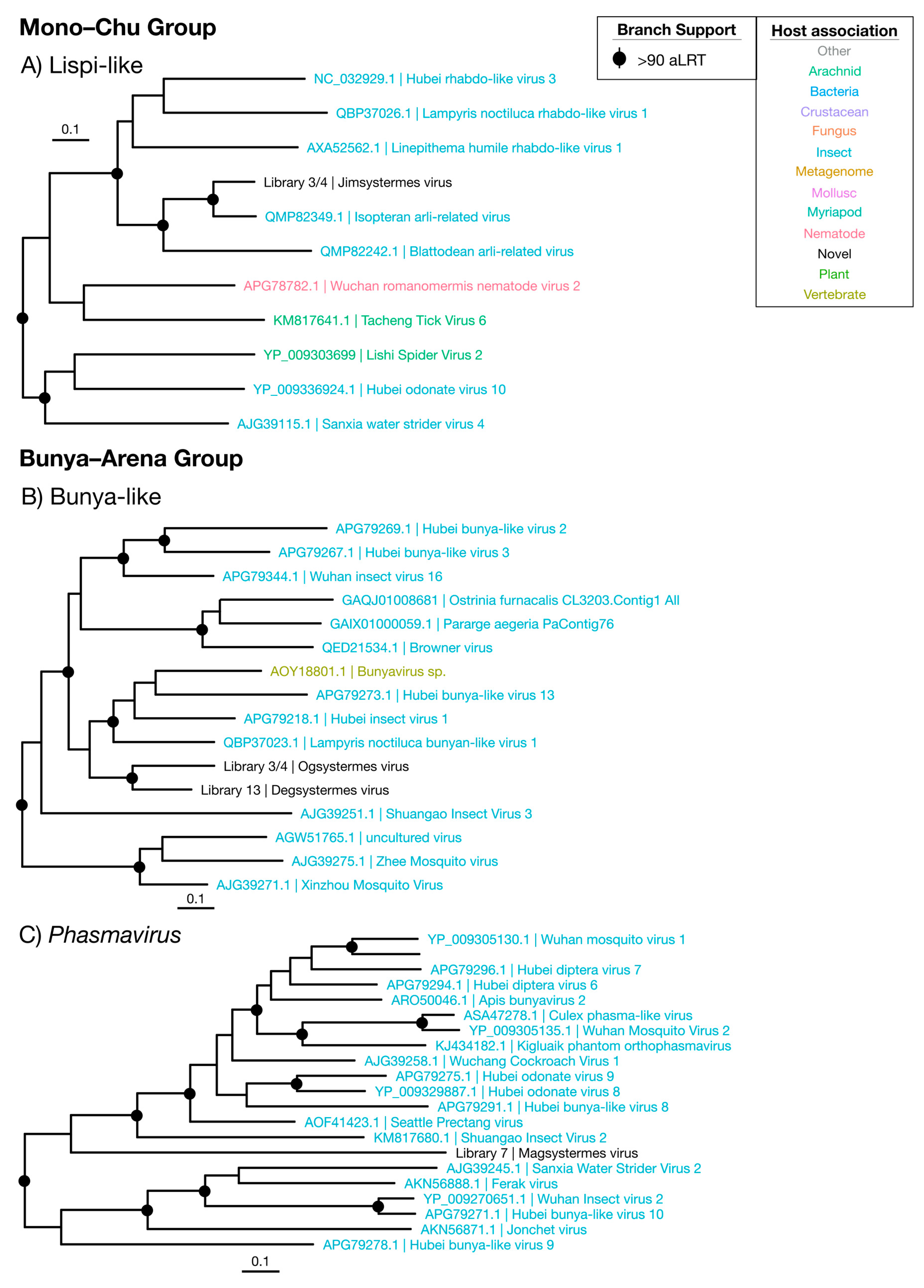
3.4.1. Mono–Chu Viruses
3.4.2. Bunya–Arena Viruses
3.5. Double-Stranded RNA Viruses
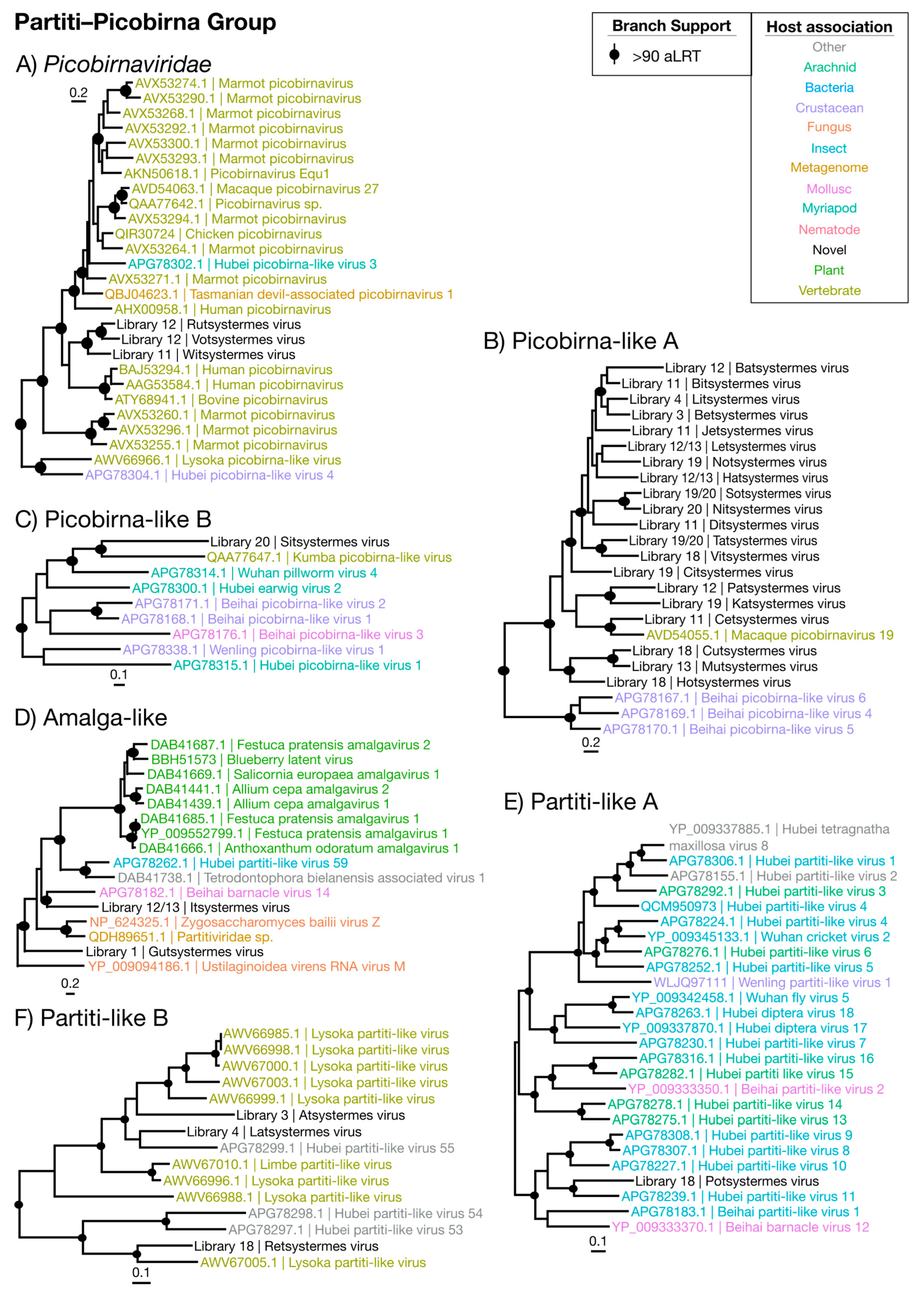
Partiti–Picobirna Viruses
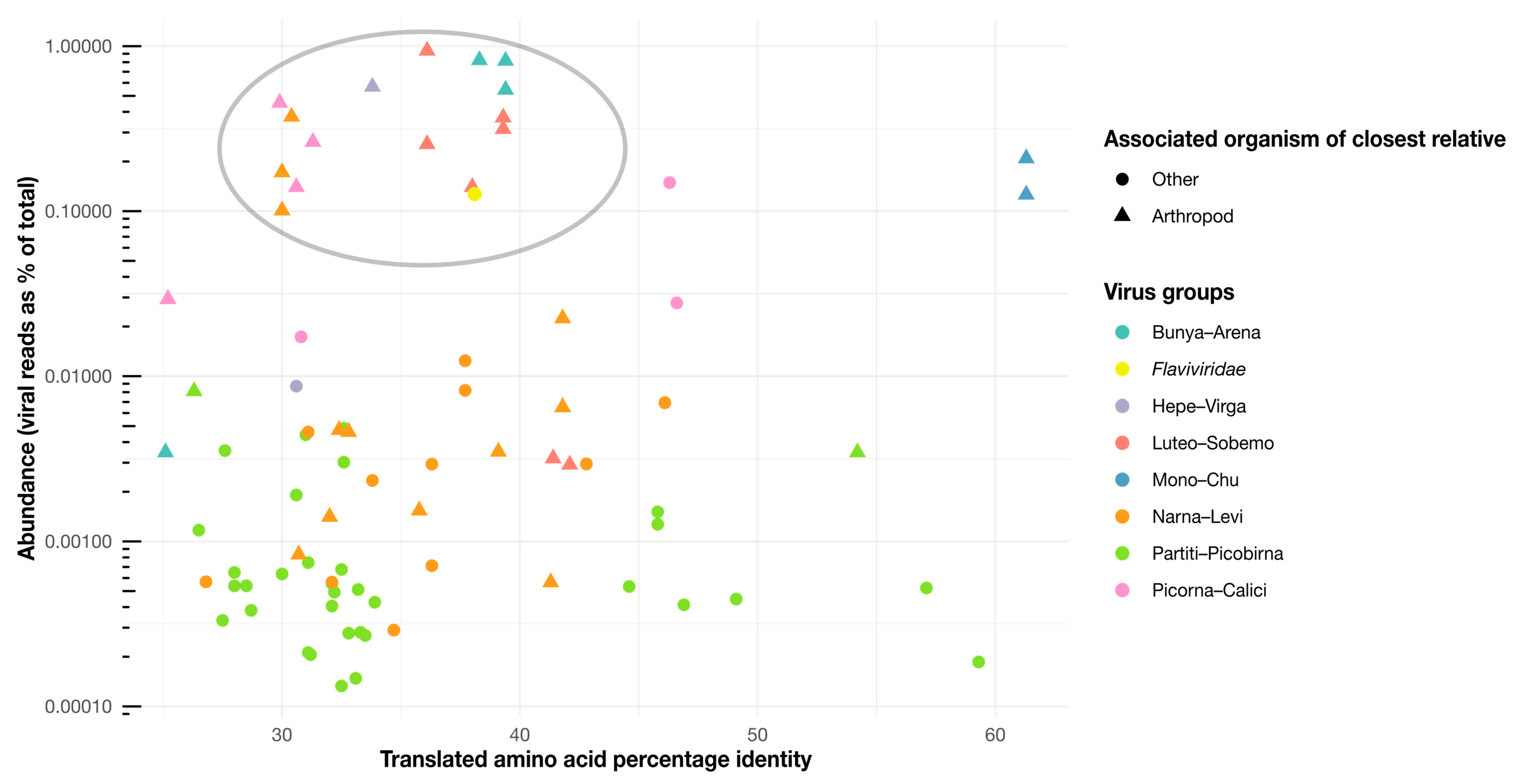
3.6. Correlates of RNA Virome Diversity and Abundance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mora, C.; Tittensor, D.P.; Adl, S.; Simpson, A.G.; Worm, B. How many species are there on earth and in the ocean? PLoS Biol. 2011, 9, e1001127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RefSeq Genome Database. Available online: https://ncbi.nlm.nih.gov/genome/viruses/ (accessed on 23 August 2020).
- Li, C.-X.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Kang, Y.-J.; Chen, L.-J.; Qin, X.-C.; Xu, J.; Holmes, E.C.; Zhang, Y.-Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539. [Google Scholar] [CrossRef] [PubMed]
- Krishna, K.; Grimaldi, D.A.; Krishna, V.; Engel, M.S. Treatise on the isoptera of the world: Termitidae. Bull. Am. Mus. Nat. Hist. 2013, 523, 973–1495. [Google Scholar] [CrossRef]
- Brune, A. Symbiotic digestion of lignocellulose in termite guts. Nat. Rev. Micro. 2014, 12, 168. [Google Scholar] [CrossRef]
- Bucek, A.; Šobotník, J.; He, S.; Shi, M.; Mcmahon, D.P.; Holmes, E.C.; Roisin, Y.; Lo, N.; Bourguignon, T. Evolution of termite symbiosis informed by transcriptome–based phylogenies. Curr. Biol. 2019, 29, 3728–3734. [Google Scholar] [CrossRef]
- Tikhe, C.V.; Husseneder, C. Metavirome sequencing of the termite gut reveals the presence of an unexplored bacteriophage community. Front. Microbiol. 2018, 8, 2548. [Google Scholar] [CrossRef] [Green Version]
- Altizer, S.; Nunn, C.L.; Thrall, P.H.; Gittleman, J.L.; Antonovics, J.; Cunningham, A.A.; Dobson, A.P.; Ezenwa, V.; Jones, K.E.; Pedersen, A.B.; et al. Social organization and parasite risk in mammals: Integrating theory and empirical studies. Annu. Rev. Ecol. Evol. Syst. 2003, 34, 517–547. [Google Scholar] [CrossRef] [Green Version]
- Pie, M.R.; Rosengaus, R.B.; Traniello, J.F. Nest architecture, activity pattern, worker density and the dynamics of disease transmission in social insects. J. Theol. Biol. 2004, 226, 45–51. [Google Scholar] [CrossRef]
- Manley, R.; Boots, M.; Wilfert, L. Review: Emerging viral disease risk to pollinating insects: Ecological, evolutionary and anthropogenic factors. J. Appl. Ecol. 2015, 52, 331–340. [Google Scholar] [CrossRef]
- Schoonvaere, K.; Smagghe, G.; Francis, F.; de Graaf, D.C. Study of the metatranscriptome of eight social and solitary wild bee species reveals novel viruses and bee parasites. Front. Microbiol. 2018, 9, 177. [Google Scholar] [CrossRef] [PubMed]
- Beaurepaire, A.; Piot, N.; Doublet, V.; Antunez, K.; Campbell, E.; Chantawannakul, P.; Chejanovsky, N.; Gajda, A.; Heerman, M.; Panziera, D.; et al. Diversity and global distribution of viruses of the western honey bee Apis mellifera. Insects 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Remnant, E.J.; Shi, M.; Buchmann, G.; Blacquière, T.; Holmes, E.C.; Beekman, M.; Ashe, A. A diverse range of novel RNA viruses in geographically distinct honey bee populations. J. Virol. 2017, 91, e00158-e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanez, O.; Piot, N.; Dalmon, A.; de Miranda, J.; Chantawannakul, P.; Panziera, D.; Amiri, E.; Smagghe, G.; Schroeder, D.; Chejanovsky, N. Bee viruses: Routes of infection in hymenoptera. Front. Microbiol. 2020, 11, 943. [Google Scholar] [CrossRef]
- Chouvenc, T.; Mullins, A.J.; Efstathion, C.A.; Su, N.-Y. Virus-like symptoms in a termite (isoptera: Kalotermitidae) field colony. Fla. Entomol. 2013, 96, 1612–1614. [Google Scholar] [CrossRef]
- Levin, D.B.; Adachi, D.; Williams, L.L.; Myles, T.G. Host specificity and molecular characterization of the Entomopoxvirus of the lesser migratory grasshopper, Melanoplus sanguinipes. J. Invertebr. Pathol. 1993, 62, 241–247. [Google Scholar] [CrossRef]
- Al Fazairy, A.A.; Hassan, F.A. Infection of termites by spodoptera littoralis nuclear polyhedrosis virus. Int. J. Trop. Insect Sci. 1988, 9, 37–39. [Google Scholar] [CrossRef]
- Pramono, A.K.; Kuwahara, H.; Itoh, T.; Toyoda, A.; Yamada, A.; Hongoh, Y. Discovery and complete genome sequence of a bacteriophage from an obligate intracellular symbiont of a cellulolytic protist in the termite gut. Microbes Environ. 2017, 32, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Rosario, K.; Mettel, K.A.; Benner, B.E.; Johnson, R.; Scott, C.; Yusseff-Vanegas, S.Z.; Baker, C.C.M.; Cassill, D.L.; Storer, C.; Varsani, A.; et al. Virus discovery in all three major lineages of terrestrial arthropods highlights the diversity of single-stranded DNA viruses associated with invertebrates. PeerJ 2018, 6, e5761. [Google Scholar] [CrossRef]
- Kerr, M.; Rosario, K.; Baker, C.C.M.; Breitbart, M. Discovery of four novel circular single-stranded DNA viruses in fungus-farming termites. Genome Announc. 2018, 6, e00318-18. [Google Scholar] [CrossRef] [Green Version]
- Käfer, S.; Paraskevopoulou, S.; Zirkel, F.; Wieseke, N.; Donath, A.; Petersen, M.; Jones, T.C.; Liu, S.; Zhou, X.; Middendorf, M.; et al. Re-assessing the diversity of negative strand RNA viruses in insects. PLoS Pathog. 2019, 15, e1008224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapelinskaya, T.V.; Martynova, E.U.; Korolev, A.L.; Schal, C.; Mukha, D.V. Transcription of the German cockroach densovirus BgDNV genome: Alternative processing of viral RNAs. Dokl. Biochem. Biophys. 2008, 421, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Mukha, D.V.; Chumachenko, A.G.; Dykstra, M.J.; Kurtti, T.J.; Schal, C. Characterization of a new densovirus infecting the German cockroach, Blattella germanica. J. Gen. Virol. 2006, 87, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. Blast+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2018, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Lam, T.T.; Zhu, H.; Guan, Y. Two methods for mapping and visualizing associated data on phylogeny using ggtree. Mol. Biol. Evol. 2018, 35, 3041–3043. [Google Scholar] [CrossRef] [PubMed]
- Revell, L.J. Phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 7 October 2020).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Hothorn, T.; Bretz, F.; Westfall, P. Simultaneous inference in general parametric models. Biom. J. 2008, 50, 346–363. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 7 October 2020).
- Harvey, E.; Rose, K.; Eden, J.-S.; Lo, N.; Abeyasuriya, T.; Shi, M.; Doggett, S.L.; Holmes, E.C. Extensive diversity of RNA viruses in australian ticks. J. Virol. 2019, 93, e01358-18. [Google Scholar] [CrossRef] [Green Version]
- Olendraite, I.; Brown, K.; Valles, S.M.; Firth, A.E.; Chen, Y.; Guérin, D.M.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E.; et al. ICTV virus taxonomy profile: Polycipiviridae. J. Gen. Virol. 2019, 100, 554. [Google Scholar] [CrossRef]
- Shackelton, L.A.; Holmes, E.C. The role of alternative genetic codes in viral evolution and emergence. J. Theor. Biol. 2008, 254, 128–134. [Google Scholar] [CrossRef]
- Nibert, M.L.; Vong, M.; Fugate, K.K.; Debat, H.J. Evidence for contemporary plant mitoviruses. Virology 2018, 518, 14–24. [Google Scholar] [CrossRef]
- Maes, P.; Adkins, S.; Alkhovsky, S.V.; Avšič-Županc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, É.; Blair, C.D.; Briese, T.; et al. Taxonomy of the order bunyavirales: Second update 2018. Arch. Virol. 2019, 164, 927–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yinda, C.K.; Vanhulle, E.; Conceição-Neto, N.; Beller, L.; Deboutte, W.; Shi, C.; Ghogomu, S.M.; Maes, P.; Van Ranst, M.; Matthijnssens, J. Gut virome analysis of cameroonians reveals high diversity of enteric viruses, including potential interspecies transmitted viruses. mSphere 2019, 4, e00585-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, S.R.; Wang, D. Extensive conservation of prokaryotic ribosomal binding sites in known and novel picobirnaviruses. Virology 2018, 516, 108–114. [Google Scholar] [CrossRef]
- Viljakainen, L.; Borshagovski, A.M.; Saarenpää, S.; Kaitala, A.; Jurvansuu, J. Identification and characterisation of common glow-worm RNA viruses. Virus Genes 2020, 56, 236–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, E.; Rose, K.; Eden, J.-S.; Lawrence, A.; Doggett, S.L.; Holmes, E.C. Identification of diverse arthropod associated viruses in native australian fleas. Virology 2019, 535, 189–199. [Google Scholar] [CrossRef]
- Debat, H.J. An RNA virome associated to the golden orb-weaver spider Nephila clavipes. Front. Microbiol. 2017, 8, 2097. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Shen, D.; Zhou, F.; Wang, G.; An, C. Identification of immunity-related genes in Ostrinia furnacalis against entomopathogenic fungi by RNA-seq analysis. PLoS ONE 2014, 9, e86436. [Google Scholar]
- Carter, J.M.; Baker, S.C.; Pink, R.; Carter, D.R.; Collins, A.; Tomlin, J.; Gibbs, M.; Breuker, C.J. Unscrambling butterfly oogenesis. BMC Genom. 2013, 14, 283. [Google Scholar] [CrossRef] [Green Version]
- Martínez, L.C.; Masachessi, G.; Carruyo, G.; Ferreyra, L.J.; Barril, P.A.; Isa, M.B.; Giordano, M.O.; Ludert, J.E.; Nates, S.V. Picobirnavirus causes persistent infection in pigs. Infect. Genet. Evol. 2010, 10, 984–988. [Google Scholar] [CrossRef]
- Starr, E.P.; Nuccio, E.E.; Pett-Ridge, J.; Banfield, J.F.; Firestone, M.K. Metatranscriptomic reconstruction reveals RNA viruses with the potential to shape carbon cycling in soil. Proc. Natl. Acad. Sci. USA 2019, 116, 25900–25908. [Google Scholar] [CrossRef] [Green Version]
- Parry, R.; Asgari, S. Discovery of novel crustacean and cephalopod flaviviruses: Insights into the evolution and circulation of flaviviruses between marine invertebrate and vertebrate Hosts. J. Virol. 2019, 93, e00432-e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourguignon, T.; Lo, N.; Dietrich, C.; Šobotník, J.; Sidek, S.; Roisin, Y.; Brune, A.; Evans, T.A. Rampant host switching shaped the termite gut microbiome. Curr. Biol. 2018, 28, 649–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Termite Species | Termite Family | Country | Diet | Procedure | Library | SRA Accession |
|---|---|---|---|---|---|---|
| Basidentitermes aurivillii | Termitidae | Kenya | Soil | Head-only | 1 | SRX6715944 |
| Amitermes sp. | Termitidae | Australia | Wood | Head-only | 2 | SRX6715975 |
| Occasitermes sp.1 | Termitidae | Australia | Wood | Whole-body | 3 1 | SRR8924823 |
| Occasitermes sp.1 | Termitidae | Australia | Wood | Whole-body | 4 1 | SRR8924822 |
| Constrictotermes cavifrons | Termitidae | French Guiana | Lichen | Head-only | 5 | SRX6715954 |
| Cephalotermes rectangularis | Termitidae | Cameroon | Wood | Head-only | 6 | SRX6715969 |
| Labiotermes labralis | Termitidae | French Guiana | Soil | Head-only | 7 | SRX6715938 |
| Cornitermes pugnax | Termitidae | French Guiana | Wood | Head-only | 8 | SRX6715963 |
| Embiratermes neotenicus | Termitidae | French Guiana | Soil | Head-only | 9 | SRX6715941 |
| Microcerotermes sp | Termitidae | French Guiana | Wood | Head-only | 10 | SRX6715964 |
| Coptotermes acinaciformis | Rhinotermitidae | Australia | Wood | Whole-body | 11 | SRR8924826 |
| Heterotermes ferox | Rhinotermitidae | Australia | Wood | Whole-body | 12 1 | SRR8924824 |
| Heterotermes ferox | Rhinotermitidae | Australia | Wood | Whole-body | 13 1 | SRR8924825 |
| Schedorhinotermes intermedius | Rhinotermitidae | Australia | Wood | Whole-body | 14 1 | SRR8924829 |
| Schedorhinotermes intermedius | Rhinotermitidae | Australia | Wood | Whole-body | 15 1 | SRR8924828 |
| Termitogeton planus | Rhinotermitidae | West Papua, Indonesia | Wood | Head-only | 16 | SRX6715976 |
| Epicalotermes kempae | Kalotermitidae | Kenya | Wood | Head-only | 17 | SRX6715943 |
| Cryptotermes secundus | Kalotermitidae | Australia | Wood | Whole-body | 18 | SRR8924827 |
| Mastotermes darwiniensis | Mastotermitidae | Australia | Wood | Whole-body | 19 2 | SRR8924831 |
| Mastotermes darwiniensis | Mastotermitidae | Australia | Wood | Whole-body | 20 2 | SRR8924830 |
| Clade | Figure | Group | Novel Viruses | Host Prediction | Mean Abundance | Mean % Identity |
|---|---|---|---|---|---|---|
| Idaeovirus-like | Figure 5A | Hepe–Virga | 1 | Plant | 0.0087 | 30.6 |
| Negev-like | Figure 5B | Hepe–Virga | 1 | Termite | 0.57 | 33.8 |
| Sobemo-like | Figure 5C | Luteo–Sobemo | 5 | Termite | 0.29 | 38.9 |
| Iflaviridae | Figure 6B | Picorna–Calici | 2 | Termite | 0.20 | 31.0 |
| Polycipiviridae | Figure 6C | Picorna–Calici | 2 | Termite | 0.065 | 38.8 |
| Nora-like | Figure 6D | Picorna–Calici | 1 | Termite | 0.45 | 29.9 |
| Kelp fly virus related | Figure 6E | Picorna–Calici | 1 | Termite | 0.029 | 24.5 |
| Flaviviridae | Figure 7A | Flaviviridae | 1 | Termite | 0.13 | 38.1 |
| Mitovirus(-like) | Figure 7B | Narna–Levi | 15 | Symbiont/No inference | 0.025 | 34.1 |
| Narna-like | Figure 7C | Narna–Levi | 1 | No inference | 0.14 | 30.0 |
| Leviviridae | Figure 7D | Narna–Levi | 3 | Bacteria | 0.0015 | 33.9 |
| Lispi-like | Figure 8A | Mono–Chu | 1 | Termite | 0.17 | 36.1 |
| Bunya-like | Figure 8B | Bunya–Arena | 2 | Termite | 0.73 | 39.0 |
| Phasmavirus | Figure 8C | Bunya–Arena | 1 | Termite | 0.0035 | 25.1 |
| Picobirnaviridae | Figure 9A | Partiti–Picobirna | 3 | Symbiont | 0.0011 | 46.2 |
| Picobirna-like A | Figure 9B | Partiti–Picobirna | 20 | Symbiont | 0.00097 | 32.0 |
| Picobirna-like B | Figure 9C | Partiti–Picobirna | 1 | Symbiont | 0.0036 | 27.6 |
| Amalga-like | Figure 9D | Partiti–Picobirna | 2 | Symbiont | 0.0030 | 28.3 |
| Partiti-like A | Figure 9E | Partiti–Picobirna | 1 | Symbiont | 0.0035 | 54.2 |
| Partiti-like B | Figure 9F | Partiti–Picobirna | 3 | Symbiont | 0.00041 | 53.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Lay, C.; Shi, M.; Buček, A.; Bourguignon, T.; Lo, N.; Holmes, E.C. Unmapped RNA Virus Diversity in Termites and Their Symbionts. Viruses 2020, 12, 1145. https://doi.org/10.3390/v12101145
Le Lay C, Shi M, Buček A, Bourguignon T, Lo N, Holmes EC. Unmapped RNA Virus Diversity in Termites and Their Symbionts. Viruses. 2020; 12(10):1145. https://doi.org/10.3390/v12101145
Chicago/Turabian StyleLe Lay, Callum, Mang Shi, Aleš Buček, Thomas Bourguignon, Nathan Lo, and Edward C. Holmes. 2020. "Unmapped RNA Virus Diversity in Termites and Their Symbionts" Viruses 12, no. 10: 1145. https://doi.org/10.3390/v12101145
APA StyleLe Lay, C., Shi, M., Buček, A., Bourguignon, T., Lo, N., & Holmes, E. C. (2020). Unmapped RNA Virus Diversity in Termites and Their Symbionts. Viruses, 12(10), 1145. https://doi.org/10.3390/v12101145