Metagenomic Approach with the NetoVIR Enrichment Protocol Reveals Virus Diversity within Ethiopian Honey Bees (Apis mellifera simensis)

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Area
2.2. Sample Collection and Transportation
2.3. Homogenization, Centrifugation and Filtration
2.4. Nuclease Treatment and Nucleic Acid Extraction
2.5. cDNA Synthesis, Amplification and Purification
2.6. Data Processing
2.7. Data Accessibility
2.8. Virus Quantification and Detection Using RT-qPCR
2.9. Phylogenetic Analysis
2.10. Negative Strand Detection Using RT-MLPA
3. Results
3.1. High-Level Taxonomic Classification
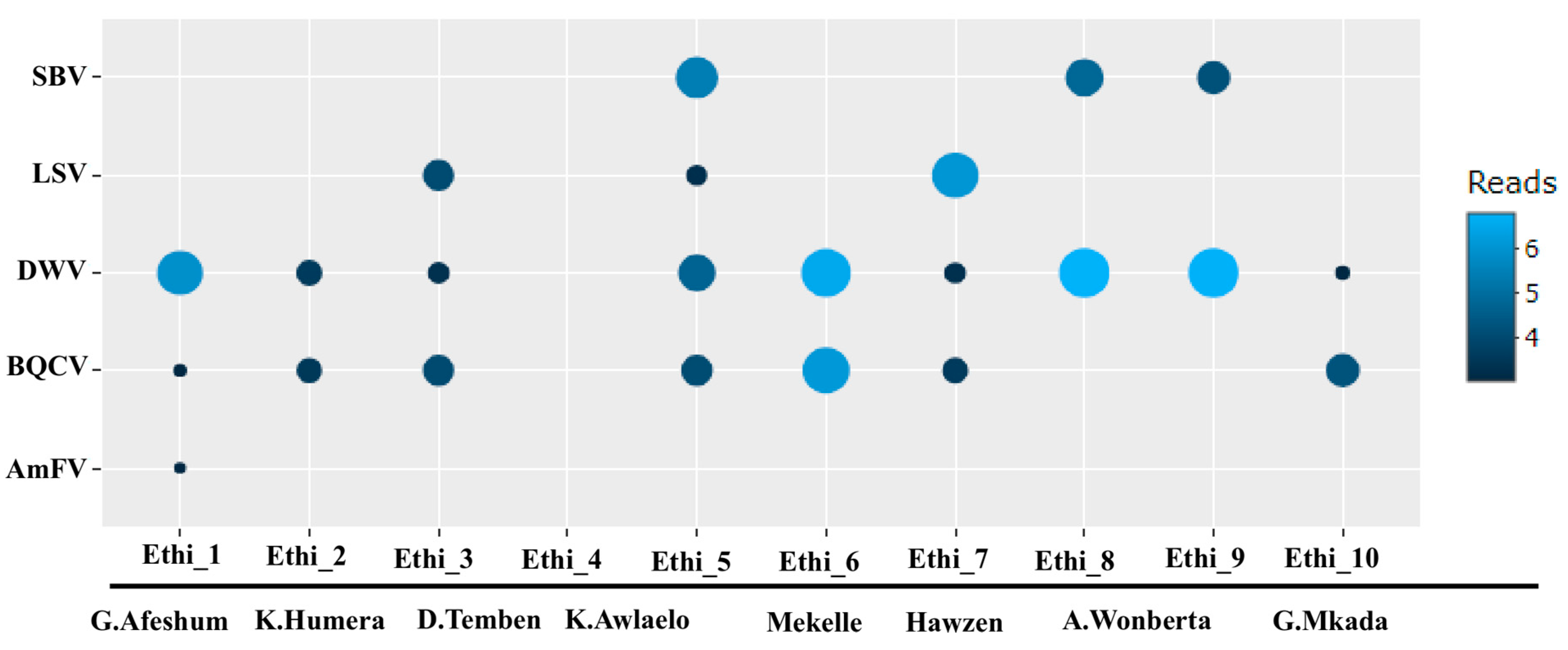
3.2. Presence of Known Honey Bee Viruses
3.3. Atypical Viruses
3.4. Virus Quantification
3.5. Phylogenetic Analysis
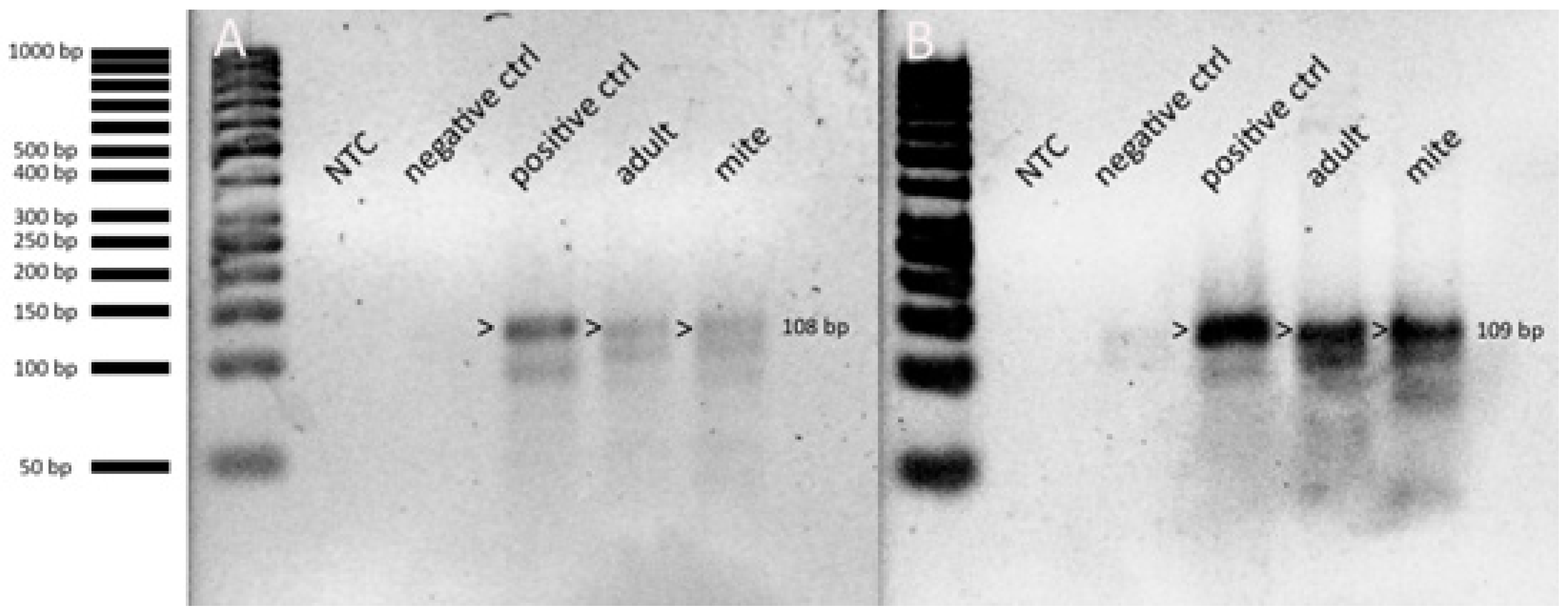
3.6. LJV and KiV Transmission and Replication
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gallai, N.; Salles, J.-M.; Settele, J.; Vaissière, B.E. Economic valuation of the vulnerability of world agriculture confronted with pollinator decline. Ecol. Econ. 2009, 68, 810–821. [Google Scholar] [CrossRef]
- Calderone, N.W. Insect Pollinated Crops, Insect Pollinators and US Agriculture: Trend Analysis of Aggregate Data for the Period 1992–2009. PLoS ONE 2012, 7, e37235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.M.; Vaissiere, B.E.; Cane, J.H.; Steffan-Dewenter, I.; Cunningham, S.A.; Kremen, C.; Tscharntke, T. Importance of pollinators in changing landscapes for world crops. Proc. R. Soc. B Boil. Sci. 2007, 274, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, A.; Brodschneider, R.; Adjlane, N.; Ballis, A.; Brusbardis, V.; Charrire, J.D.; Chlebo, R.; Coffey, M.F.; Cornelissen, B.; da Costa, C.A.; et al. Loss rates of honey bee colonies during winter 2017/18 in 36 countries participating in the COLOSS survey, including effects of forage sources. J. Apic. Res. 2019, 58, 479–485. [Google Scholar] [CrossRef] [Green Version]
- VanEngelsdorp, D.; Caron, D.; Hayes, J.; Underwood, R.; Rennich, K.; Spleen, A.; Andree, M.; Snyder, R.; Lee, K.; Roccasecca, K.; et al. A national survey of managed honey bee 2010—11 winter colony losses in the USA: Results from the Bee Informed Partnership. J. Apic. Res. 2012, 51, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.M.; Loh, E.H.; Rostal, M.K.; Zambrana-Torrelio, C.M.; Mendiola, L.; Daszak, P. Pathogens, Pests, and Economics: Drivers of Honey Bee Colony Declines and Losses. Ecohealth 2013, 10, 434–445. [Google Scholar] [CrossRef]
- Yalçınkaya, A.; Keskin, N. The investigation of honey bee diseases after colony losses in Hatay and Adana provinces of Turkey. Mellifera 2010, 10, 24–31. [Google Scholar]
- Ellis, J.D.; Munn, P.A. The worldwide health status of honey bees. Bee World 2005, 86, 88–101. [Google Scholar] [CrossRef]
- VanEngelsdorp, D.; Hayes, J.H.; Underwood, R.M.; Pettis, J. A survey of honey bee colony losses in the U.S. fall 2007 to spring 2008. PLoS ONE 2008, 3, e4071. [Google Scholar] [CrossRef]
- Ravoet, J.; Maharramov, J.; Meeus, I.; De Smet, L.; Wenseleers, T.; Smagghe, G.; de Graaf, D.C. Comprehensive Bee Pathogen Screening in Belgium Reveals Crithidia mellificae as a New Contributory Factor to Winter Mortality. PLoS ONE 2013, 8, e72443. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, S.D.; Ochoa, R.; Bauchan, G.; Gulbronson, C.; Mowery, J.D.; Cohen, A.; Lim, D.; Joklik, J.; Cicero, J.M.; Ellis, J.D.; et al. Varroa destructor feeds primarily on honey bee fat body tissue and not hemolymph. Proc. Natl. Acad. Sci. USA 2019, 116, 1792–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Prisco, G.; Annoscia, D.; Margiotta, M.; Ferrara, R.; Varricchio, P.; Zanni, V.; Caprio, E.; Nazzi, F.; Pennacchio, F. A mutualistic symbiosis between a parasitic mite and a pathogenic virus undermines honey bee immunity and health. Proc. Natl. Acad. Sci. USA 2016, 113, 3203–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowen-Walker, P.L.; Martin, S.J.; Gunn, A. The transmission of deformed wing virus between honeybees (Apis mellifera L.) by the ectoparasitic mite Varroa jacobsoni Oud. J. Invertebr. Pathol. 1999, 73, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Mockel, N.; Gisder, S.; Genersch, E. Horizontal transmission of deformed wing virus: Pathological consequences in adult bees (Apis mellifera) depend on the transmission route. J. Gen. Virol. 2011, 92, 370–377. [Google Scholar] [CrossRef]
- Posada-Florez, F.; Childers, A.K.; Heerman, M.C.; Egekwu, N.I.; Cook, S.C.; Chen, Y.P.; Evans, J.D.; Ryabov, E.V. Deformed wing virus type A, a major honey bee pathogen, is vectored by the mite Varroa destructor in a non-propagative manner. Sci. Rep. UK 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.Q.; Yang, X.L.; Cox-Foster, D.; Cui, L.W. The role of varroa mites in infections of Kashmir bee virus (KBV) and deformed wing virus (DWV) in honey bees. Virology 2005, 342, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.J.; Highfield, A.C.; Brettell, L.; Villalobos, E.M.; Budge, G.E.; Powell, M.; Nikaido, S.; Schroeder, D.C. Global Honey Bee Viral Landscape Altered by a Parasitic Mite. Science 2012, 336, 1304–1306. [Google Scholar] [CrossRef] [PubMed]
- Gisder, S.; Aumeier, P.; Genersch, E. J. Gen. Virol. 2009, 90, 463–467. [CrossRef]
- De Miranda, J.R.; Cordoni, G.; Budge, G. The Acute bee paralysis virus-Kashmir bee virus-Israeli acute paralysis virus complex. J. Invertebr. Pathol. 2010, 103, S30–S47. [Google Scholar] [CrossRef]
- Genersch, E.; Aubert, M. Emerging and re-emerging viruses of the honey bee (Apis mellifera L.). Vet. Res. 2010, 41, 54. [Google Scholar] [CrossRef] [Green Version]
- Grozinger, C.M.; Flenniken, M.L. Bee Viruses: Ecology, Pathogenicity, and Impacts. Annu. Rev. Entomol. 2019, 64, 205–226. [Google Scholar] [CrossRef] [PubMed]
- Steinhauer, N.; Kulhanek, K.; Antunez, K.; Human, H.; Chantawannakul, P.; Chauzat, M.P.; vanEngelsdorp, D. Drivers of colony losses. Curr. Opin. Insect. Sci. 2018, 26, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.M.; Nielsen, S.L.; Kryger, P. Varroa-Virus Interaction in Collapsing Honey Bee Colonies. PLoS ONE 2013, 8, e57540. [Google Scholar] [CrossRef] [Green Version]
- Kajobe, R.; Marris, G.; Budge, G.; Laurenson, L.; Cordoni, G.; Jones, B.; Wilkins, S.; Cuthbertson, A.G.S.; Brown, M.A. First molecular detection of a viral pathogen in Ugandan honey bees. J. Invertebr. Pathol. 2010, 104, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Amakpe, F.; De Smet, L.; Brunain, M.; Ravoet, J.; Jacobs, F.J.; Reybroeck, W.; Sinsin, B.; de Graaf, D.C. Discovery of Lake Sinai virus and an unusual strain of acute bee paralysis virus in West African apiaries. Apidologie 2016, 47, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Muli, E.; Patch, H.; Frazier, M.; Frazier, J.; Torto, B.; Baumgarten, T.; Kilonzo, J.; Kimani, J.N.; Mumoki, F.; Masiga, D.; et al. Evaluation of the distribution and impacts of parasites, pathogens, and pesticides on honey bee (Apis mellifera) populations in east Africa. PLoS ONE 2014, 9, e94459. [Google Scholar] [CrossRef] [PubMed]
- Ongus, J.R.; Fombong, A.T.; Irungu, J.; Masiga, D.; Raina, S. Prevalence of common honey bee pathogens at selected apiaries in Kenya, 2013/2014. Int. J. Trop Insect. Sci. 2018, 38, 58–70. [Google Scholar] [CrossRef]
- Mumoki, F.N.; Fombong, A.; Muli, E.; Muigai, W.T.; Masiga, D. An inventory of documented diseases of African honeybees. Afr. Entomol. 2014, 22, 473–487. [Google Scholar] [CrossRef]
- Pirk, C.W.W.; Strauss, U.; Yusuf, A.A.; Demares, F.; Human, H. Honeybee health in Africa-a review. Apidologie 2016, 47, 276–300. [Google Scholar] [CrossRef]
- Haddad, N.J.; Noureddine, A.; Al-Shagour, B.; Loucif-Ayad, W.; El-Niweiri, M.A.A.; Anaswah, E.; Abu Hammour, W.; El-Obeid, D.; Imad, A.; Shebl, M.A.; et al. Distribution and variability of deformed wing virus of honeybees (Apis mellifera) in the Middle East and North Africa. Insect Sci. 2017, 24, 103–113. [Google Scholar] [CrossRef]
- Galbraith, D.A.; Fuller, Z.L.; Ray, A.M.; Brockmann, A.; Frazier, M.; Gikungu, M.W.; Martinez, J.F.I.; Kapheim, K.M.; Kerby, J.T.; Kocher, S.D.; et al. Investigating the viral ecology of global bee communities with high-throughput metagenomics. Sci. Rep. UK 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remnant, E.J.; Shi, M.; Buchmann, G.; Blacquiere, T.; Holmes, E.C.; Beekman, M.; Ashe, A. A Diverse Range of Novel RNA Viruses in Geographically Distinct Honey Bee Populations. J. Virol. 2017, 91, e00158-17. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.M.K.; Anderson, D.L.; Durr, P.A. Metagenomic analysis of Varroa-free Australian honey bees (Apis mellifera) shows a diverse Picornavirales virome. J. Gen. Virol. 2018, 99, 818–826. [Google Scholar] [CrossRef]
- Alger, S.A.; Burnham, P.A.; Boncristiani, H.F.; Brody, A.K. RNA virus spillover from managed honeybees (Apis mellifera) to wild bumblebees (Bombus spp.). PLoS ONE 2019, 14, e0217822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, H.; Brown, M.J.F.; Stevenson, P.C. The role of disease in bee foraging ecology. Curr. Opin. Insect Sci. 2017, 21, 60–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aizenberg-Gershtein, Y.; Izhaki, I.; Halpern, M. Do Honeybees Shape the Bacterial Community Composition in Floral Nectar? PLoS ONE 2013, 8, e67556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, K.; Stenchly, K.; Coulibaly, D.; Pauly, A.; Dimobe, K.; Steffan-Dewenter, I.; Konate, S.; Goetze, D.; Porembski, S.; Linsenmair, K.E. Impact of human disturbance on bee pollinator communities in savanna and agricultural sites in Burkina Faso, West Africa. Ecol. Evol. 2018, 8, 6827–6838. [Google Scholar] [CrossRef] [PubMed]
- Granberg, F.; Vicente-Rubiano, M.; Rubio-Guerri, C.; Karlsson, O.E.; Kukielka, D.; Belak, S.; Sanchez-Vizcaino, J.M. Metagenomic Detection of Viral Pathogens in Spanish Honeybees: Co-Infection by Aphid Lethal Paralysis, Israel Acute Paralysis and Lake Sinai Viruses. PLoS ONE 2013, 8, e57459. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, D.M.; Mushegian, A.R.; Dolja, V.V.; Koonin, E.V. New dimensions of the virus world discovered through metagenomics. Trends Microbiol. 2010, 18, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Deboutte, W.; Beller, L.; Yinda, C.K.; Maes, P.; de Graaf, D.C.; Matthijnssens, J. Honey-bee-associated prokaryotic viral communities reveal wide viral diversity and a profound metabolic coding potential. Proc. Natl. Acad. Sci. USA 2020, 117, 10511–10519. [Google Scholar] [CrossRef]
- Schoonvaere, K.; De Smet, L.; Smagghe, G.; Vierstraete, A.; Braeckman, B.P.; de Graaf, D.C. Unbiased RNA Shotgun Metagenomics in Social and Solitary Wild Bees Detects Associations with Eukaryote Parasites and New Viruses. PLoS ONE 2016, 11, e0168456. [Google Scholar] [CrossRef] [PubMed]
- Schoonvaere, K.; Smagghe, G.; Francis, F.; de Graaf, D.C. Study of the Metatranscriptome of Eight Social and Solitary Wild Bee Species Reveals Novel Viruses and Bee Parasites. Front. Microbiol. 2018, 9, 177. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.J.; Wang, J.; Todd, A.K.; Bissielo, A.B.; Yen, S.H.; Strydom, H.; Moore, N.E.; Ren, X.Y.; Huang, Q.S.; Carter, P.E.; et al. Evaluation of rapid and simple techniques for the enrichment of viruses prior to metagenomic virus discovery. J. Virol. Methods 2014, 195, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conceicao-Neto, N.; Zeller, M.; Lefrere, H.; De Bruyn, P.; Beller, L.; Deboutte, W.; Yinda, C.K.; Lavigne, R.; Maes, P.; Van Ranst, M.; et al. Modular approach to customise sample preparation procedures for viral metagenomics: A reproducible protocol for virome analysis. Sci. Rep. UK 2015, 5, 16532. [Google Scholar] [CrossRef] [Green Version]
- Conceicao-Neto, N.; Yinda, K.C.; Van Ranst, M.; Matthijnssens, J. NetoVIR: Modular Approach to Customize Sample Preparation Procedures for Viral Metagenomics. Methods Mol. Biol. 2018, 1838, 85–95. [Google Scholar]
- de Miranda, J.R.; Bailey, L.; Ball, B.V.; Blanchard, P.; Budge, G.E.; Chejanovsky, N.; Chen, Y.P.; Gauthier, L.; Genersch, E.; de Graaf, D.C.; et al. Standard methods for virus research in Apis mellifera. J. Apic. Res. 2013, 52, 1–56. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsgren, E.; de Miranda, J.R.; Isaksson, M.; Wei, S.; Fries, I. Deformed wing virus associated with Tropilaelaps mercedesae infesting European honey bees (Apis mellifera). Exp. Appl. Acarol. 2009, 47, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Locke, B.; Forsgren, E.; Fries, I.; de Miranda, J.R. Acaricide Treatment Affects Viral Dynamics in Varroa destructor-Infested Honey Bee Colonies via both Host Physiology and Mite Control (vol 78, pg 227, 2012). Appl. Environ. Microb. 2012, 78, 2073. [Google Scholar] [CrossRef] [Green Version]
- Schurr, F.; Tison, A.; Militano, L.; Cheviron, N.; Sircoulomb, F.; Riviere, M.P.; Ribiere-Chabert, M.; Thiery, R.; Dubois, E. Validation of quantitative real-time RT-PCR assays for the detection of six honeybee viruses. J. Virol. Methods 2019, 270, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.C.; Smith, D.K.; Zhu, H.C.; Guan, Y.; Lam, T.T.Y. GGTREE: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- De Smet, L.; Ravoet, J.; de Miranda, J.R.; Wenseleers, T.; Mueller, M.Y.; Moritz, R.F.A.; de Graaf, D.C. BeeDoctor, a Versatile MLPA-Based Diagnostic Tool for Screening Bee Viruses. PLoS ONE 2012, 7, e47953. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.L.; Waldron, F.M.; Robertson, S.; Crowson, D.; Ferrari, G.; Quintana, J.F.; Brouqui, J.M.; Bayne, E.H.; Longdon, B.; Buck, A.H.; et al. The Discovery, Distribution, and Evolution of Viruses Associated with Drosophila melanogaster. PLoS Biol. 2015, 13, e1002210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuiskunen, A.; Leparc-Goffart, I.; Boubis, L.; Monteil, V.; Klingstrom, J.; Tolou, H.J.; Lundkvist, A.; Plumet, S. Self-priming of reverse transcriptase impairs strand-specific detection of dengue virus RNA. J. Gen. Virol. 2010, 91, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Begna, D. Occurrences and distributions of honeybee (Apis mellifera Jemenetica) varroa mite (Varroa destructor) in Tigray Region, Ethiopia. J. Fish. Livest. Prod. 2014, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Begna, D.; Gela, A.; Negera, T.; Bezabeh, A. Identifying the species, effects and seasonal dynamics of honeybee varroa mites: A newly emerging parasite to Ethiopian honeybee. Int. J. Sci. Res. Environ. Sci. Toxicol. 2016, 1, 4. [Google Scholar] [CrossRef]
- Gebremedhn, H.; Amssalu, B.; De Smet, L.; de Graaf, D.C. Factors restraining the population growth of Varroa destructor in Ethiopian honey bees (Apis mellifera simensis). PLoS ONE 2019, 14, e0223236. [Google Scholar] [CrossRef] [Green Version]
- O’Flaherty, B.M.; Li, Y.; Tao, Y.; Paden, C.R.; Queen, K.; Zhang, J.; Dinwiddie, D.L.; Gross, S.M.; Schroth, G.P.; Tong, S.X. Comprehensive viral enrichment enables sensitive respiratory virus genomic identification and analysis by next generation sequencing. Genome Res. 2018, 28, 869–877. [Google Scholar] [CrossRef] [Green Version]
- Bailey, L.; Carpenter, J.M.; Woods, R.D. Properties of a Filamentous Virus of the Honey Bee (Apis-Mellifera). Virology 1981, 114, 1–7. [Google Scholar] [CrossRef]
- Allen, M.; Ball, B. The incidence and world distribution of honey bee viruses. Bee World 1996, 77, 141–162. [Google Scholar] [CrossRef]
- Haddad, N.; Horth, L.; Al-Shagour, B.; Adjlane, N.; Loucif-Ayad, W. Next-generation sequence data demonstrate several pathogenic bee viruses in Middle East and African honey bee subspecies (Apis mellifera syriaca, Apis mellifera intermissa) as well as their cohabiting pathogenic mites (Varroa destructor). Virus Genes 2018, 54, 694–705. [Google Scholar] [CrossRef]
- Levin, S.; Sela, N.; Erez, T.; Nestel, D.; Pettis, J.; Neumann, P.; Chejanovsky, N. New Viruses from the Ectoparasite Mite Varroa destructor Infesting Apis mellifera and Apis cerana. Viruses 2019, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ongus, J.R.; Peters, D.; Bonmatin, J.M.; Bengsch, E.; Vlak, J.M.; van Oers, M.M. Complete sequence of a picorna-like virus of the genus Iflavirus replicating in the mite Varroa destructor. J. Gen. Virol. 2004, 85, 3747–3755. [Google Scholar] [CrossRef]
- Zioni, N.; Soroker, V.; Chejanovsky, N. Replication of Varroa destructor virus 1 (VDV-1) and a Varroa destructor virus 1-deformed wing virus recombinant (VDV-1-DWV) in the head of the honey bee. Virology 2011, 417, 106–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mordecai, G.J.; Wilfert, L.; Martin, S.J.; Jones, I.M.; Schroeder, D.C. Diversity in a honey bee pathogen: First report of a third master variant of the Deformed Wing Virus quasispecies. ISME J. 2016, 10, 1264–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaurepaire, A.; Piot, N.; Doublet, V.; Antunez, K.; Campbell, E.; Chantawannakul, P.; Chejanovsky, N.; Gajda, A.; Heerman, M.; Panziera, D.; et al. Diversity and Global Distribution of Viruses of the Western Honey Bee, Apis mellifera. Insects 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- McMahon, D.P.; Natsopoulou, M.E.; Doublet, V.; Furst, M.; Weging, S.; Brown, M.J.F.; Gogol-Doring, A.; Paxton, R.J. Elevated virulence of an emerging viral genotype as a driver of honeybee loss. Proc. R. Soc. B Boil. Sci. 2016, 283, 20160811. [Google Scholar] [CrossRef]
- Kevill, J.L.; de Souza, F.S.; Sharples, C.; Oliver, R.; Schroeder, D.C.; Martin, S.J. DWV-A Lethal to Honey Bees (Apis mellifera): A Colony Level Survey of DWV Variants (A, B, and C) in England, Wales, and 32 States across the US. Viruses 2019, 11, 426. [Google Scholar] [CrossRef] [Green Version]
- Francis, R.M.; Nielsen, S.L.; Kryger, P. Patterns of viral infection in honey bee queens. J. Gen. Virol. 2013, 94, 668–676. [Google Scholar] [CrossRef]
- Amiri, E.; Meixner, M.; Nielsen, S.L.; Kryger, P. Four Categories of Viral Infection Describe the Health Status of Honey Bee Colonies. PLoS ONE 2015, 10, e0140272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaduri, S.; Stephan, J.G.; de Miranda, J.R.; Locke, B. Disentangling host-parasite-pathogen interactions in a varroa-resistant honeybee population reveals virus tolerance as an independent, naturally adapted survival mechanism. Sci. Rep. UK 2019, 9, 6221. [Google Scholar] [CrossRef] [PubMed]
- Locke, B.; Forsgren, E.; de Miranda, J.R. Increased Tolerance and Resistance to Virus Infections: A Possible Factor in the Survival of Varroa destructor-Resistant Honey Bees (Apis mellifera). PLoS ONE 2014, 9, e99998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raberg, L.; Graham, A.L.; Read, A.F. Decomposing health: Tolerance and resistance to parasites in animals. Philos. Trans. R. Soc. B Biol. Sci. 2008, 364, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Nibert, M.L.; Ghabrial, S.A.; Maiss, E.; Lesker, T.; Vainio, E.J.; Jiang, D.H.; Suzuki, N. Taxonomic reorganization of family Partitiviridae and other recent progress in partitivirus research. Virus Res. 2014, 188, 128–141. [Google Scholar] [CrossRef]
- Vainio, E.J.; Chiba, S.; Ghabrial, S.A.; Maiss, E.; Roossinck, M.; Sabanadzovic, S.; Suzuki, N.; Xie, J.T.; Nibert, M.; Consortium, I.R. ICTV Virus Taxonomy Profile: Partitiviridae. J. Gen. Virol. 2018, 99, 17–18. [Google Scholar] [CrossRef]
- Li, J.L.; Cornman, R.S.; Evans, J.D.; Pettis, J.S.; Zhao, Y.; Murphy, C.; Peng, W.J.; Wu, J.; Hamilton, M.; Boncristiani, H.F.; et al. Systemic Spread and Propagation of a Plant-Pathogenic Virus in European Honeybees, Apis mellifera. mBio 2014, 5, e00898-13. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.M.K.; Ireland, K.B.; Tay, W.T.; Paini, D. Honey bee-assisted surveillance for early plant virus detection. Ann. Appl. Biol. 2018, 173, 285–293. [Google Scholar] [CrossRef]
- Darzi, E.; Smith, E.; Shargil, D.; Lachman, O.; Ganot, L.; Dombrovsky, A. The honeybee Apis mellifera contributes to Cucumber green mottle mosaic virus spread via pollination. Plant Pathol. 2018, 67, 244–251. [Google Scholar] [CrossRef]
- Boccardo, G.; Lisa, V.; Luisoni, E.; Milne, R.G. Cryptic Plant-Viruses. Adv. Virus Res. 1987, 32, 171–214. [Google Scholar]
- Khetarpal, R.K.; Maury, Y. Pea Seed-Borne Mosaic-Virus—A Review. Agronomie 1987, 7, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Blawid, R.; Stephan, D.; Maiss, E. Molecular characterization and detection of Vicia cryptic virus in different Vicia faba cultivars. Arch. Virol. 2007, 152, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Card, S.D.; Pearson, M.N.; Clover, G.R.G. Plant pathogens transmitted by pollen. Australas. Plant Path. 2007, 36, 455–461. [Google Scholar] [CrossRef]
- Wylie, K.M.; Wylie, T.N.; Buller, R.; Herter, B.; Cannella, M.T.; Storch, G.A. Detection of Viruses in Clinical Samples by Use of Metagenomic Sequencing and Targeted Sequence Capture. J. Clin. Microbiol. 2018, 56, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Levitt, A.L.; Rajotte, E.G.; Holmes, E.C.; Ostiguy, N.; Vanengelsdorp, D.; Lipkin, W.I.; Depamphilis, C.W.; Toth, A.L.; Cox-Foster, D.L. RNA Viruses in Hymenopteran Pollinators: Evidence of Inter-Taxa Virus Transmission via Pollen and Potential Impact on Non-Apis Hymenopteran Species. PLoS ONE 2010, 5, e14357. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.L.; Longdon, B.; Lewis, S.H.; Obbard, D.J. Twenty-Five New Viruses Associated with the Drosophilidae (Diptera). Evol. Bioinform. 2016, 12, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrau, T.; Hiebert, N.; Vilcinskas, A.; Lee, K.Z. Identification and characterization of natural viruses associated with the invasive insect pest Drosophila suzukii. J. Invertebr. Pathol. 2018, 154, 74–78. [Google Scholar] [CrossRef]
- Bachmann, P.A.; Hoggan, M.D.; Kurstak, E.; Melnick, J.L.; Pereira, H.G.; Tattersall, P.; Vago, C. Parvoviridae—2nd Report. Intervirology 1979, 11, 248–254. [Google Scholar] [CrossRef]
- Ravoet, J.; De Smet, L.; Meeus, I.; Smagghe, G.; Wenseleers, T.; de Graaf, D.C. Widespread occurrence of honey bee pathogens in solitary bees. J. Invertebr. Pathol. 2014, 122, 55–58. [Google Scholar] [CrossRef]
- Antunez, K.; Anido, M.; Branchiccela, B.; Harriet, J.; Campa, J.; Invernizzi, C.; Santos, E.; Higes, M.; Martin-Hernandez, R.; Zunino, P. Seasonal Variation of Honeybee Pathogens and its Association with Pollen Diversity in Uruguay. Microb. Ecol. 2015, 70, 522–533. [Google Scholar] [CrossRef]
- Genersch, E.; von der Ohe, W.; Kaatz, H.; Schroeder, A.; Otten, C.; Buchler, R.; Berg, S.; Ritter, W.; Muhlen, W.; Gisder, S.; et al. The German bee monitoring project: A long term study to understand periodically high winter losses of honey bee colonies. Apidologie 2010, 41, 332–352. [Google Scholar] [CrossRef] [Green Version]
- Schouten, J.P.; McElgunn, C.J.; Waaijer, R.; Zwijnenburg, D.; Diepvens, F.; Pals, G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002, 30, 57. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gebremedhn, H.; Deboutte, W.; Schoonvaere, K.; Demaeght, P.; De Smet, L.; Amssalu, B.; Matthijnssens, J.; de Graaf, D.C. Metagenomic Approach with the NetoVIR Enrichment Protocol Reveals Virus Diversity within Ethiopian Honey Bees (Apis mellifera simensis). Viruses 2020, 12, 1218. https://doi.org/10.3390/v12111218
Gebremedhn H, Deboutte W, Schoonvaere K, Demaeght P, De Smet L, Amssalu B, Matthijnssens J, de Graaf DC. Metagenomic Approach with the NetoVIR Enrichment Protocol Reveals Virus Diversity within Ethiopian Honey Bees (Apis mellifera simensis). Viruses. 2020; 12(11):1218. https://doi.org/10.3390/v12111218
Chicago/Turabian StyleGebremedhn, Haftom, Ward Deboutte, Karel Schoonvaere, Peter Demaeght, Lina De Smet, Bezabeh Amssalu, Jelle Matthijnssens, and Dirk C. de Graaf. 2020. "Metagenomic Approach with the NetoVIR Enrichment Protocol Reveals Virus Diversity within Ethiopian Honey Bees (Apis mellifera simensis)" Viruses 12, no. 11: 1218. https://doi.org/10.3390/v12111218
APA StyleGebremedhn, H., Deboutte, W., Schoonvaere, K., Demaeght, P., De Smet, L., Amssalu, B., Matthijnssens, J., & de Graaf, D. C. (2020). Metagenomic Approach with the NetoVIR Enrichment Protocol Reveals Virus Diversity within Ethiopian Honey Bees (Apis mellifera simensis). Viruses, 12(11), 1218. https://doi.org/10.3390/v12111218