Influenza A Virus Inhibits RSV Infection via a Two-Wave Expression of IFIT Proteins

Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Samples and Ethical Approval
2.2. Viral RNA Extraction
2.3. Co-Infection of HEp2 Cells with Influenza A/H3N2 and RSV
2.4. Co-Infection of Mice with Influenza A/PR/8/1934 and RSV
2.5. Mass Spectrometry
2.6. Genes Silencing of IFIT1, IFIT2, IFIT3 and IFI44
- IFIT1
- forward, 5′-TCTCAGAGGAGCCTGGCTAA-3′
- reverse, 5′-TCAGGCATTTCATCGTCATC-3′
- IFIT2
- forward, 5′-CGAACAGCTGAGAATTGCAC-3′
- reverse, 5′-TGCACATTGTGGCTTTGAAT-3′
- IFIT3
- forward, 5′-CGGAACAGCAGAGACACAGA-3′
- reverse, 5′-CTGCCTCGTTGTTACCATCT-3’
- IFI44
- forward, 5′- AGCTGGGAAGTCCAGCTTTT-3′
- reverse, 5′-CCCCAGTGAGTCACACAGAA-3′
2.7. Gene Overexpression of IFIT1, IFIT2, IFIT3, and IFI44
- IFIT1
- forward, 5′-TTTTCTCGAGGCCGCCACCATGAGTACAAATGGTGATGATC-3′
- reverse, 5′-AAAGCTAGCCTAAGGACCTTGTCTCACAGAGTTC-3′
- IFIT2
- forward, 5′-TTTTCTCGAGGCCGCCACCATGAGTGAGAACAATAAGAA-3′
- reverse, 5′-AAAGCTAGCTCATTCCCCATTCCAGCTTGATGCT-3′
- IFIT3
- forward, 5′-TTTTCTCGAGGCCGCCACCATGAGTGAGGTCACCAAGAATTC-3′
- reverse, 5′-AAAGCTAGCTCAGTTCAGTTGCTCTGAGTTAG-3′
- IFI44
- forward, 5′-TTTTCTCGAGGCCGCCACCATGGCAGTGACAACTCGTTTGAC-3′
- reverse, 5′-CCCGCTAGCCTACCCGCTAGCCTATTTTTTTCCTTGTGCACAGTTGATAATTTCCTCCCTTAGATTC-3′
2.8. Western Blot
2.9. Infection of IFIT-Silenced/Overexpressed HEp2 Cells with RSV
2.10. Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
- Mice lung:
- Ifit1
- forward, 5′-TGCTGAGATGGACTGTGAGG-3′
- reverse, 5′-GTGCCAATTCTTGCACATTG-3′
- ifit2
- forward, 5′-ACATGGGCCAGTTCTCAAAG-3′
- reverse, 5′-ATGCACATAGGCGTTGTTTG-3′
- ifit3
- forward, 5′-CTGGTCACCTGGGGAAACTA-3′
- reverse, 5′-ATCGCAGTGCTTTTCCAAGT-3′
- ifi44
- forward, 5′-GTTTGACATGGCAGCAAGAA-3′
- reverse, 5′-TATTGCGAGCAGTCAGTTGG-3′
2.11. Statistical Analysis
3. Results
3.1. Influenza Infection Inhibits RSV
3.2. Influenza Infection Inhibits RSV In Vitro
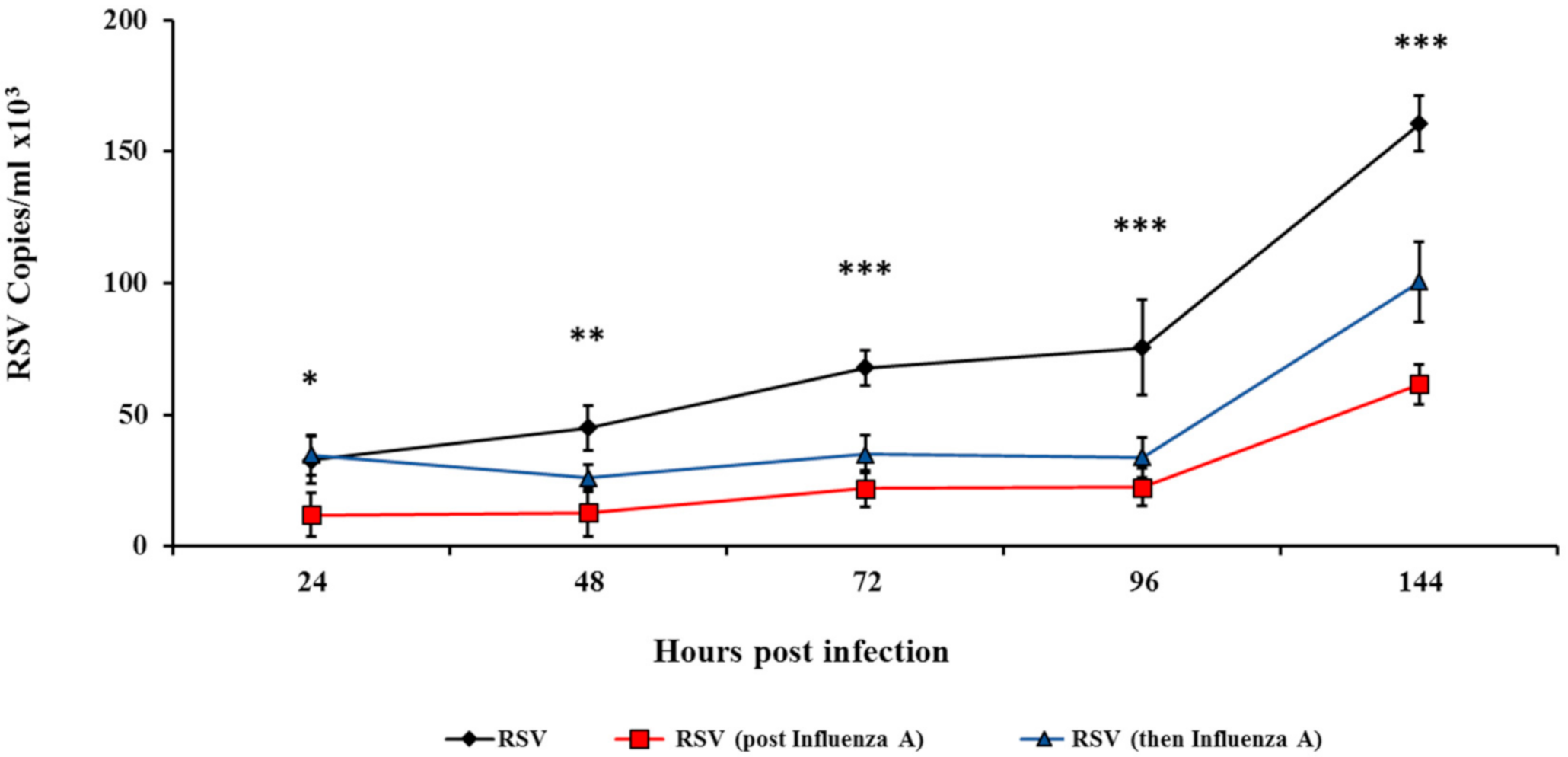
3.3. Influenza Infection Inhibits RSV In Vivo in a Two-Wave Modality
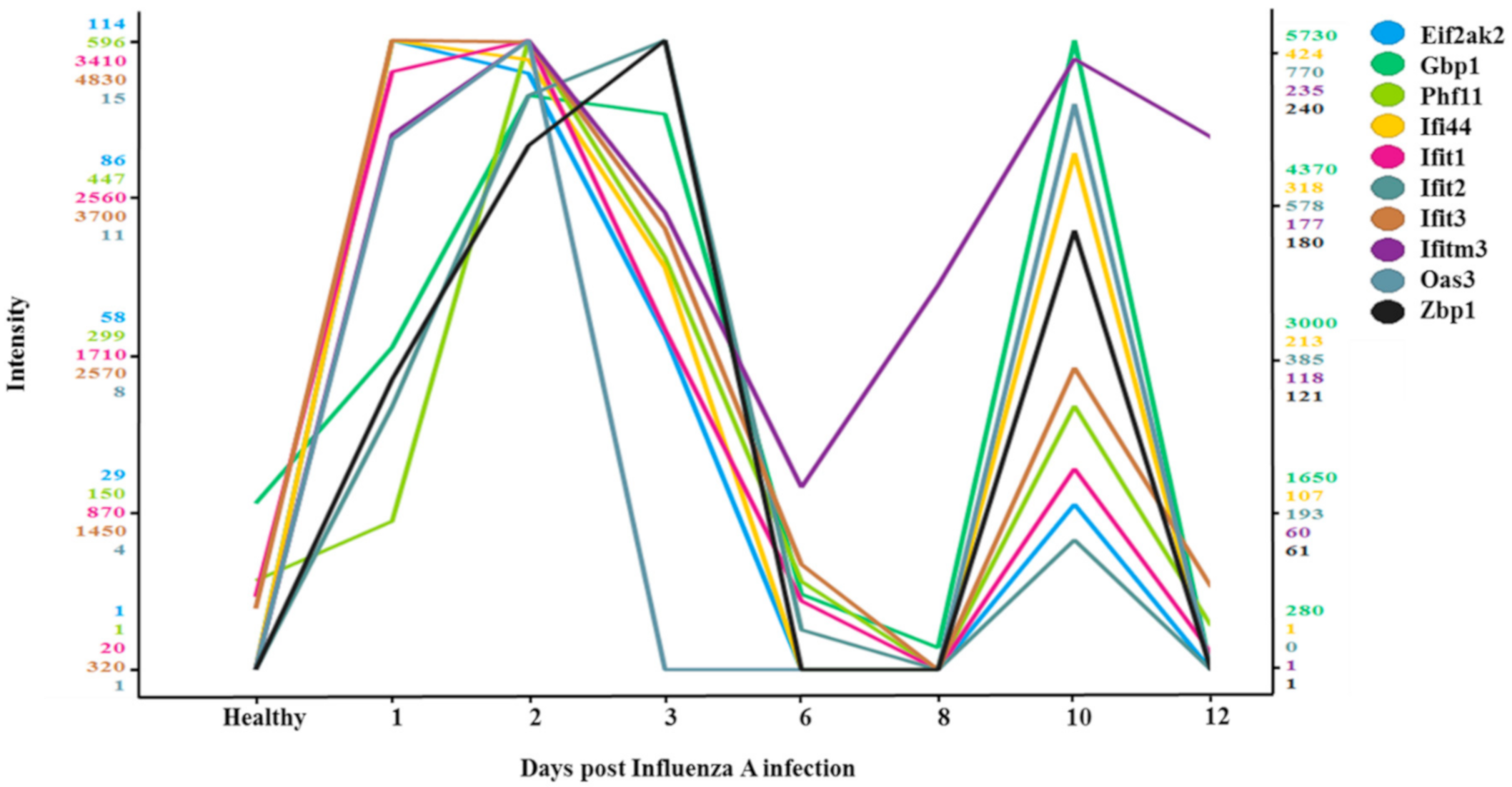
3.4. Two-Wave Elevation of Anti-Viral Proteins Following Influenza Infection
3.5. IFIT Members Inhibit RSV Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Griffin, M.R.; Coffey, C.S.; Neuzil, K.M.; Mitchel, E.F.; Wright, P.F.; Edwards, K.M. Winter viruses: Influenza- and respiratory syncytial virus-related morbidity in chronic lung disease. Arch. Intern. Med. 2002, 162, 1229–1236. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xu, B.; Guo, J.; Li, C.; An, S.; Zhou, Y.; Chen, A.; Deng, L.; Fu, Z.; Zhu, Y.; et al. Genetic variations in the fusion protein of respiratory syncytial virus isolated from children hospitalized with community-acquired pneumonia in China. Sci. Rep. 2018, 8, 4491. [Google Scholar] [CrossRef] [PubMed]
- Vangeti, S.; Yu, M.; Smed-Sörensen, A. Respiratory Mononuclear Phagocytes in Human Influenza A Virus Infection: Their Role in Immune Protection and As Targets of the Virus. Front. Immunol. 2018, 9, 1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, J.B.; Rojas-Rueda, D.; Alonso, J.; Antó, J.M.; Cardona, P.-J.; Fernández, E.; Garcia-Basteiro, A.L.; Benavides, F.G.; Glenn, S.D.; Krish, V.; et al. La carga de enfermedad en España: Resultados del Estudio de la Carga Global de las Enfermedades 2016. Med. Clínica 2018, 151, 171–190. [Google Scholar] [CrossRef] [PubMed]
- Yaron-Yakoby, H.; Sefty, H.; Pando, R.; Dichtiar, R.; Katz, M.A.; Stein, Y.; Mandelboim, M.; Mendelson, E.; Shohat, T.; Glatman-Freedman, A.; et al. Effectiveness of influenza vaccine in preventing medically-attended influenza virus infection in primary care, Israel, influenza seasons 2014/15 and 2015/16. Eurosurveillance 2018, 23, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widjaja, I.; Wicht, O.; Luytjes, W.; Leenhouts, K.; Rottier, P.J.M.; Van Kuppeveld, F.J.M.; Haijema, B.J.; De Haan, C.A.M. Characterization of Epitope-Specific Anti-Respiratory Syncytial Virus (Anti-RSV) Antibody Responses after Natural Infection and after Vaccination with Formalin-Inactivated RSV. J. Virol. 2016, 90, 5965–5977. [Google Scholar] [CrossRef] [Green Version]
- Taubenberger, J.K.; Kash, J.C. Influenza Virus Evolution, Host Adaptation, and Pandemic Formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Hirsh, S.; Hindiyeh, M.; Kolet, L.; Regev, L.; Sherbany, H.; Yaary, K.; Mendelson, E.; Mandelboim, M. Epidemiological Changes of Respiratory Syncytial Virus (RSV) Infections in Israel. PLoS ONE 2014, 9, e90515. [Google Scholar] [CrossRef]
- Míguez, A.; Iftimi, A.; Montes, F. Temporal association between the influenza virus and respiratory syncytial virus (RSV): RSV as a predictor of seasonal influenza. Epidemiol. Infect. 2016, 144, 2621–2632. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Michal, J.J.; Zhang, L.; Ding, B.; Lunney, J.K.; Liu, B.; Jiang, Z. Interferon Induced IFIT Family Genes in Host Antiviral Defense. Int. J. Biol. Sci. 2013, 9, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Daugherty, M.D.; Schaller, A.M.; Geballe, A.P.; Malik, H.S. Evolution-guided functional analyses reveal diverse antiviral specificities encoded by IFIT1 genes in mammals. eLife 2016, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, M.A.G.; Ribaudo, M.; Guo, J.-T.; Barik, S. Identification of Interferon-Stimulated Gene Proteins That Inhibit Human Parainfluenza Virus Type 3. J. Virol. 2016, 90, 11145–11156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mears, H.V.; Sweeney, T.R. Better together: The role of IFIT protein–protein interactions in the antiviral response. J. Gen. Virol. 2018, 99, 1463–1477. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Sweeney, T.R.; Skabkin, M.A.; Skabkina, O.V.; Hellen, C.U.; Pestova, T.V. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1- and 5′ppp- mRNAs. Nucleic Acids Res. 2013, 42, 3228–3245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habjan, M.; Hubel, P.; Lacerda, L.; Benda, C.; Holze, C.; Eberl, C.H.; Mann, A.; Kindler, E.; Gil-Cruz, C.; Ziebuhr, J.; et al. Sequestration by IFIT1 Impairs Translation of 2′O-unmethylated Capped RNA. PLoS Pathog. 2013, 9, e1003663. [Google Scholar] [CrossRef] [Green Version]
- Fensterl, V.; Sen, G.C. Interferon-Induced Ifit Proteins: Their Role in Viral Pathogenesis. J. Virol. 2014, 89, 2462–2468. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, A.; Takahashi, K.; Okajima, A.; Kitamura, N. Induction of the human gene for p44, a hepatitis-C-associated microtubular aggregate protein, by interferon-alpha/beta. Eur. J. Biochem. 1994, 224, 877–883. [Google Scholar] [CrossRef]
- Bochkov, Y.A.; Hanson, K.M.; Keles, S.; Brockman-Schneider, R.A.; Jarjour, N.N.; Gern, J.E. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol. 2009, 3, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Kaczkowski, B.; Rossing, M.; Andersen, D.K.; Dreher, A.; Morevati, M.; Visser, M.A.; Winther, O.; Nielsen, F.C.; Norrild, B. Integrative analyses reveal novel strategies in HPV11,-16 and -45 early infection. Sci. Rep. 2012, 2, 515. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Nogales, A.; Martinez-Sobrido, L.; Topham, D.J. Interferon-Induced Protein 44 Interacts with Cellular FK506-Binding Protein 5, Negatively Regulates Host Antiviral Responses, and Supports Virus Replication. mBio 2019, 10, 8. [Google Scholar] [CrossRef] [Green Version]
- Meningher, T.; Hindiyeh, M.; Regev, L.; Sherbany, H.; Mendelson, E.; Mandelboim, M. Relationships between A(H1N1)pdm09 influenza infection and infections with other respiratory viruses. Influ. Respir. Viruses 2014, 8, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Domon, B. Mass Spectrometry and Protein Analysis. Science 2006, 312, 212–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regev, L.; Meningher, T.; Hindiyeh, M.; Mendelson, E.; Mandelboim, M. Increase Human Metapneumovirus Mediated Morbidity following Pandemic Influenza Infection. PLoS ONE 2012, 7, e34750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walzl, G.; Tafuro, S.; Moss, P.; Openshaw, P.J.; Hussell, T. Influenza Virus Lung Infection Protects from Respiratory Syncytial Virus–Induced Immunopathology. J. Exp. Med. 2000, 192, 1317–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meskill, S.D.; Revell, P.A.; Chandramohan, L.; Cruz, A.T. Prevalence of co-infection between respiratory syncytial virus and influenza in children. Am. J. Emerg. Med. 2016, 35, 495–498. [Google Scholar] [CrossRef] [PubMed]
- McCullers, J.A. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat. Rev. Genet. 2014, 12, 252–262. [Google Scholar] [CrossRef]
- Korenkov, D.; Isakova-Sivak, I.; Rudenko, L. Basics of CD8 T-cell immune responses after influenza infection and vaccination with inactivated or live attenuated influenza vaccine. Expert Rev. Vaccines 2018, 17, 977–987. [Google Scholar] [CrossRef]
- MacIntyre, C.R.; Chughtai, A.A.; Barnes, M.; Ridda, I.; Seale, H.; Toms, R.; Heywood, A. The role of pneumonia and secondary bacterial infection in fatal and serious outcomes of pandemic influenza a(H1N1)pdm09. BMC Infect. Dis. 2018, 18, 637. [Google Scholar] [CrossRef] [Green Version]
- Walaza, S.; Cohen, C.; Tempia, S.; Moyes, J.; Nguweneza, A.; Madhi, S.A.; McMorrow, M.; Cohen, A.L. Influenza and tuberculosis co-infection: A systematic review. Influ. Respir. Viruses 2019, 14, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Pichlmair, A.; Lassnig, C.; Eberle, C.-A.; Górna, M.W.; Baumann, C.L.; Burkard, T.R.; Bürckstümmer, T.; Stefanovic, A.; Krieger, S.; Bennett, K.L.; et al. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar] [CrossRef]
- Johnson, B.; VanBlargan, L.A.; Xu, W.; White, J.P.; Shan, C.; Shi, P.-Y.; Zhang, R.; Adhikari, J.; Gross, M.L.; Leung, D.W.; et al. Human IFIT3 Modulates IFIT1 RNA Binding Specificity and Protein Stability. Immunity 2018, 48, 487–499. [Google Scholar] [CrossRef] [Green Version]
- Andrejeva, J.; Norsted, H.; Habjan, M.; Thiel, V.; Goodbourn, S.; Randall, R.E. ISG56/IFIT1 is primarily responsible for interferon-induced changes to patterns of parainfluenza virus type 5 transcription and protein synthesis. J. Gen. Virol. 2013, 94, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, D.F.; Andrejeva, J.; Li, X.; Inesta-Vaquera, F.; Dong, C.; Cowling, V.H.; Goodbourn, S.; Randall, R.E. Human IFIT1 Inhibits mRNA Translation of Rubulaviruses but Not Other Members of the Paramyxoviridae Family. J. Virol. 2016, 90, 9446–9456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heim, M.H.; Thimme, R. Innate and adaptive immune responses in HCV infections. J. Hepatol. 2014, 61, S14–S25. [Google Scholar] [CrossRef] [Green Version]
- Brasier, A.R. Therapeutic targets for inflammation-mediated airway remodeling in chronic lung disease. Expert Rev. Respir. Med. 2018, 12, 931–939. [Google Scholar] [CrossRef]
- Diaz, J.; Morales-Romero, J.; Pérez-Gil, G.; Bedolla-Barajas, M.; Delgado-Figueroa, N.; García-Román, R.; López-López, O.; Bañuelos, E.; Rizada-Antel, C.; Zenteno-Cuevas, R.; et al. Viral coinfection in acute respiratory infection in Mexican children treated by the emergency service: A cross-sectional study. Ital. J. Pediatr. 2015, 41, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ayegbusi, O.T.; Ajagbe, O.A.; Afowowe, T.O.; Aransi, A.T.; Olusola, B.A.; Awogbindin, I.O.; Ogunsemowo, O.O.; Faneye, A.O.; Odaibo, G.N.; Olaleye, D.O. Virus genes and host correlates of pathology are markedly reduced during respiratory syncytial and influenza virus co-infection in BALB/c mice. Heliyon 2019, 5, e01094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grayson, M.H.; Ramos, M.S.; Rohlfing, M.M.; Kitchens, R.; Wang, H.D.; Gould, A.; Agapov, E.; Holtzman, M.J. Controls for Lung Dendritic Cell Maturation and Migration during Respiratory Viral Infection. J. Immunol. 2007, 179, 1438–1448. [Google Scholar] [CrossRef]
- Cheng, L.; Ueno, A.; Cho, S.; Im, J.S.; Golby, S.; Hou, S.; Porcelli, S.A.; Yang, Y. Efficient Activation of Vα14 Invariant NKT Cells by Foreign Lipid Antigen Is Associated with Concurrent Dendritic Cell-Specific Self Recognition. J. Immunol. 2007, 178, 2755–2762. [Google Scholar] [CrossRef] [Green Version]
- Hegde, S.; Chen, X.; Keaton, J.M.; Reddington, F.; Besra, G.S.; Gumperz, J. NKT cells direct monocytes into a DC differentiation pathway. J. Leukoc. Biol. 2007, 81, 1224–1235. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Names | Function |
|---|---|
| Eif2ak2 | Inhibits viral replication via phosphorylation of the alpha subunit of eukaryotic initiation factor. |
| Gbp1 | Promote oxidative killing and deliver antimicrobial peptides to autophagolysosomes. |
| Phf11 | Inhibitor of prototype foamy virus (PFV) replication. |
| Ifi44 | Exhibits an antiviral activity against hepatitis C virus. |
| Ifit1 | Acting as a sensor of viral single- stranded RNAs and inhibiting expression of viral messenger RNAs. |
| Ifit2 | Distinguish between self and non-self mRNAs by the host during viral infection. |
| Ifit3 | IFN-induced antiviral protein which acts as an inhibitor of viral processes, and viral replication. |
| Ifitm3 | Inhibits the entry of viruses to the host cell cytoplasm. |
| Oas3 | dsRNA-activated antiviral enzyme which plays a critical role in cellular innate antiviral response. |
| Zbp1 | Participates in the detection by the host’s innate immune system of DNA from viral. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drori, Y.; Jacob-Hirsch, J.; Pando, R.; Glatman-Freedman, A.; Friedman, N.; Mendelson, E.; Mandelboim, M. Influenza A Virus Inhibits RSV Infection via a Two-Wave Expression of IFIT Proteins. Viruses 2020, 12, 1171. https://doi.org/10.3390/v12101171
Drori Y, Jacob-Hirsch J, Pando R, Glatman-Freedman A, Friedman N, Mendelson E, Mandelboim M. Influenza A Virus Inhibits RSV Infection via a Two-Wave Expression of IFIT Proteins. Viruses. 2020; 12(10):1171. https://doi.org/10.3390/v12101171
Chicago/Turabian StyleDrori, Yaron, Jasmine Jacob-Hirsch, Rakefet Pando, Aharona Glatman-Freedman, Nehemya Friedman, Ella Mendelson, and Michal Mandelboim. 2020. "Influenza A Virus Inhibits RSV Infection via a Two-Wave Expression of IFIT Proteins" Viruses 12, no. 10: 1171. https://doi.org/10.3390/v12101171
APA StyleDrori, Y., Jacob-Hirsch, J., Pando, R., Glatman-Freedman, A., Friedman, N., Mendelson, E., & Mandelboim, M. (2020). Influenza A Virus Inhibits RSV Infection via a Two-Wave Expression of IFIT Proteins. Viruses, 12(10), 1171. https://doi.org/10.3390/v12101171