Seasonal Regime Shift in the Viral Communities of a Permafrost Thaw Lake

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Site Description
2.2. Sample Collection and Processing
2.3. Identification of Viral Reads
2.4. Diversity Analyses
2.5. Genomic Analyses
2.6. Comparison to Other Viral Communities
2.7. Data Availability
3. Results and Discussion
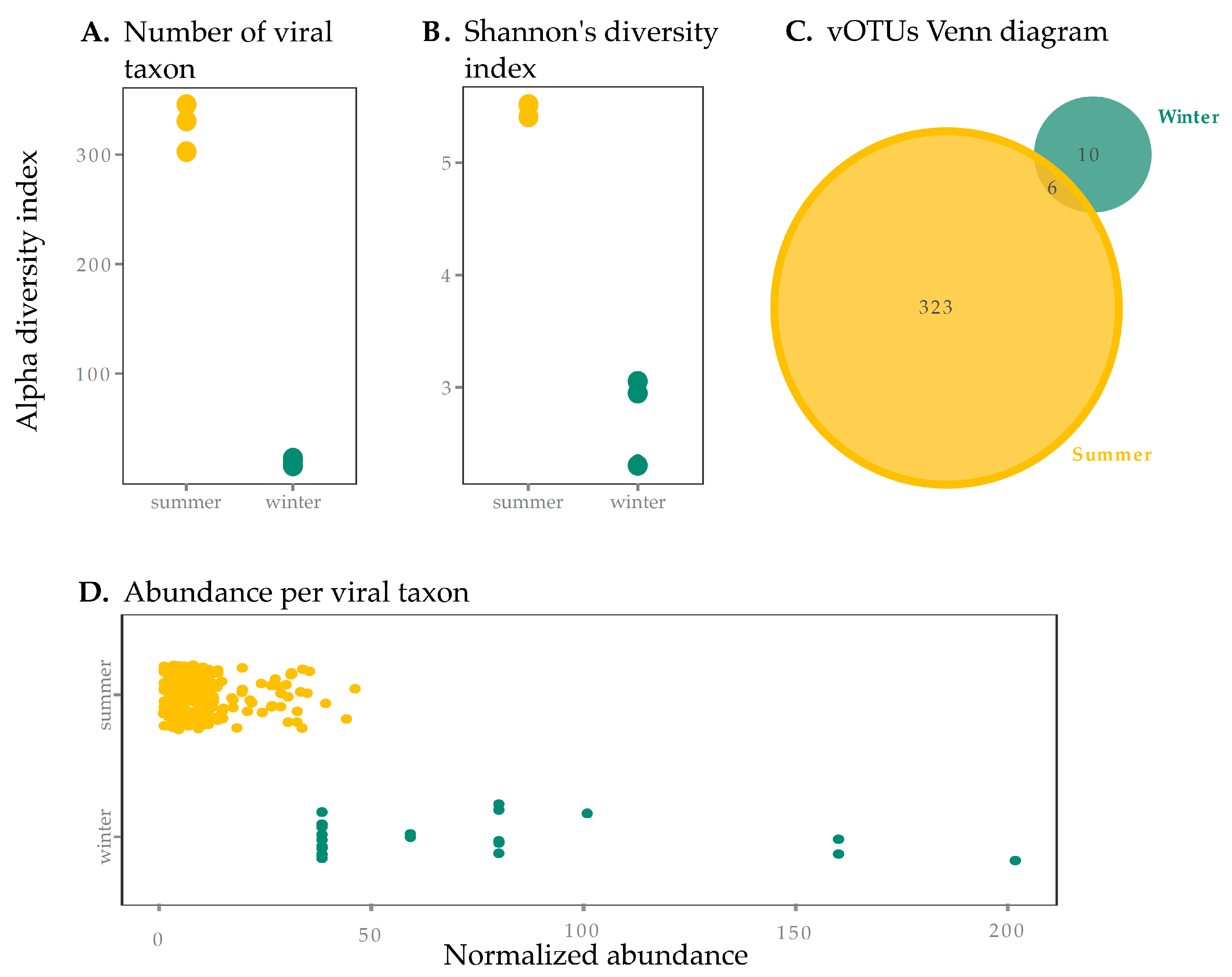
3.1. Viral Yield
3.2. Contrasting Environmental Conditions and Viral Populations Across Seasons
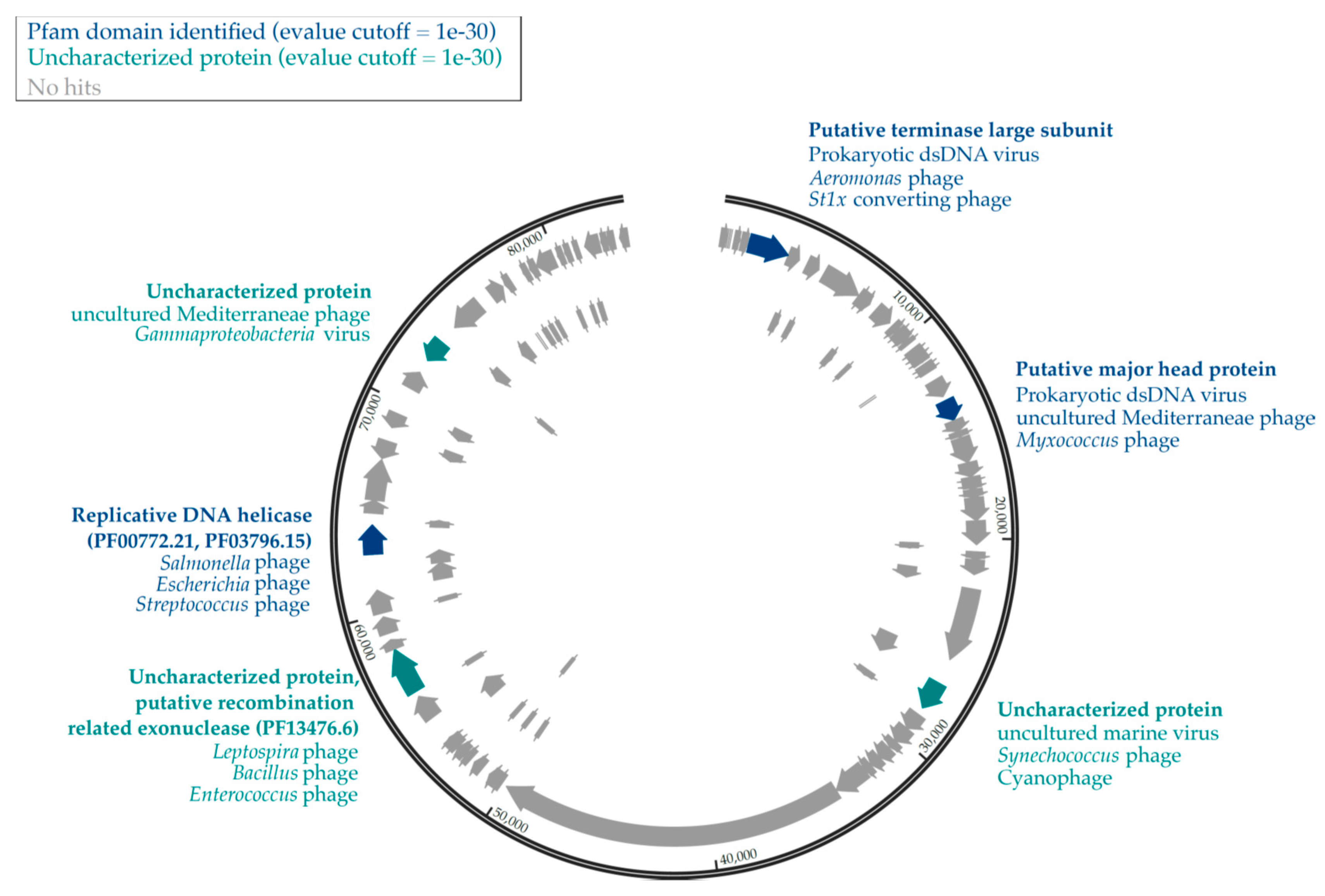
3.3. Genomic Analyses of Uncultured Viral Genomes
3.4. Comparisons to Other Sites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gruber, S. Derivation and analysis of a high-resolution estimate of global permafrost zonation. Cryosphere 2012, 6, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Schuur, E.A.G.; McGuire, A.D.; Schädel, C.; Grosse, G.; Harden, J.W.; Hayes, D.J.; Hugelius, G.; Koven, C.D.; Kuhry, P.; Lawrence, D.M.; et al. Climate change and the permafrost carbon feedback. Nature 2015, 520, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Pörtner, H.-O.; Roberts, D.C.; Masson-Delmotte, V.; Zhai, P.; Tignor, M.; Poloczanska, E.; Mintenbeck, K.; Nicolai, M.; Okem, A.; Petzold, J.; et al. IPCC. Summary for Policymakers. In IPCC Special Report on the Ocean and Cryosphere in a Changing Climate; 2019; in press. [Google Scholar]
- Biskaborn, B.K.; Smith, S.L.; Noetzli, J.; Matthes, H.; Vieira, G.; Streletskiy, D.A.; Schoeneich, P.; Romanovsky, V.E.; Lewkowicz, A.G.; Abramov, A.; et al. Permafrost is warming at a global scale. Nat. Commun. 2019, 10, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansson, J.K.; Tas, N. The microbial ecology of permafrost. Nat. Rev. Microbiol. 2014, 12, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Vonk, J.E.; Tank, S.E.; Bowden, W.B.; Laurion, I.; Vincent, W.F.; Alekseychik, P.; Amyot, M.; Billet, M.F.; Canário, J.; Cory, R.M.; et al. Reviews and syntheses: Effects of permafrost thaw on Arctic aquatic ecosystems. Biogeosciences 2015, 12, 7129–7167. [Google Scholar] [CrossRef] [Green Version]
- Pienitz, R.; Doran, P.T.; Lamoureux, S.F. Origin and Geomorphology of Lakes in the Polar Regions in: Polar Lakes and Rivers: Limnology of Arctic and Antarctic Aquatic Ecosystems; Vincent, W.F., Laybourne-Parry, J., Eds.; Oxford University Press: Oxford, UK, 2008; pp. 25–41. [Google Scholar]
- Farquharson, L.M.; Mann, D.H.; Grosse, G.; Jones, B.M.; Romanovsky, V.E. Spatial distribution of thermokarst terrain in Arctic Alaska. Geomorphology 2016, 273, 116–133. [Google Scholar] [CrossRef] [Green Version]
- Vincent, W.F.; Lemay, M.; Allard, M.; Wolfe, B.B. Adapting to permafrost change: A science framework. Eos Trans. Am. Geophys. Union 2013, 94, 373–375. [Google Scholar] [CrossRef]
- Matveev, A.; Laurion, I.; Deshpande, B.N.; Bhiry, N.; Vincent, W.F. High methane emissions from thermokarst lakes in subarctic peatlands: Methane emissions from peatland thermokarst takes. Limnol. Oceanogr. 2016, 61, S150–S164. [Google Scholar] [CrossRef] [Green Version]
- Negandhi, K.; Laurion, I.; Whiticar, M.J.; Galand, P.E.; Xu, X.; Lovejoy, C. Small thaw ponds: An unaccounted source of methane in the Canadian High Arctic. PLoS ONE 2013, 8, e78204. [Google Scholar] [CrossRef] [Green Version]
- Laurion, I.; Vincent, W.F.; MacIntyre, S.; Retamal, L.; Dupont, C.; Francus, P.; Pienitz, R. Variability in greenhouse gas emissions from permafrost thaw ponds. Limnol. Oceanogr. 2010, 55, 115–133. [Google Scholar] [CrossRef] [Green Version]
- Wik, M.; Varner, R.K.; Anthony, K.W.; MacIntyre, S.; Bastviken, D. Climate-sensitive northern lakes and ponds are critical components of methane release. Nat. Geosci. 2016, 9, 99–105. [Google Scholar] [CrossRef]
- In’t Zandt, M.H.; Liebner, S.; Welte, C.U. Roles of thermokarst lakes in a warming world. Trends Microbiol. 2020, 28, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Comte, J.; Lovejoy, C.; Crevecoeur, S.; Vincent, W.F. Co-occurrence patterns in aquatic bacterial communities across changing permafrost landscapes. Biogeosciences 2016, 13, 175–190. [Google Scholar] [CrossRef] [Green Version]
- Crevecoeur, S.; Vincent, W.F.; Comte, J.; Lovejoy, C. Bacterial community structure across environmental gradients in permafrost thaw ponds: Methanotroph-rich ecosystems. Front. Microbiol. 2015, 6, 192. [Google Scholar] [CrossRef] [Green Version]
- Vigneron, A.; Lovejoy, C.; Cruaud, P.; Kalenitchenko, D.; Culley, A.; Vincent, W.F. Contrasting winter versus summer microbial communities and metabolic functions in a permafrost thaw lake. Front. Microbiol. 2019, 10, 1656. [Google Scholar] [CrossRef] [Green Version]
- Negandhi, K.; Laurion, I.; Lovejoy, C. Bacterial communities and greenhouse gas emissions of shallow ponds in the High Arctic. Polar Biol. 2014, 37, 1669–1683. [Google Scholar] [CrossRef]
- Crevecoeur, S.; Vincent, W.F.; Lovejoy, C. Environmental selection of planktonic methanogens in permafrost thaw ponds. Sci. Rep. 2016, 6, 31312. [Google Scholar] [CrossRef] [Green Version]
- Negandhi, K.; Laurion, I.; Lovejoy, C. Temperature effects on net greenhouse gas production and bacterial communities in arctic thaw ponds. FEMS Microbiol. Ecol. 2016, 92, fiw117. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, A.E.; Howard-Varona, C.; Needham, D.M.; John, S.G.; Worden, A.Z.; Sullivan, M.B.; Waldbauer, J.R.; Coleman, M.L. Metabolic and biogeochemical consequences of viral infection in aquatic ecosystems. Nat. Rev. Microbiol. 2020, 18, 21–34. [Google Scholar] [CrossRef]
- Suttle, C.A. Environmental microbiology: Viral diversity on the global stage. Nat. Microbiol. 2016, 1, 16205. [Google Scholar] [CrossRef]
- Culley, A. New insight into the RNA aquatic virosphere via viromics. Virus Res. 2018, 244, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.S.; Rise, M.L.; Culley, A.I.; Steward, G.F. RNA viruses in the sea. FEMS Microbiol. Rev. 2009, 33, 295–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middelboe, M.; Jacquet, S.; Weinbauer, M. Viruses in freshwater ecosystems: An introduction to the exploration of viruses in new aquatic habitats. Freshw. Biol. 2008, 53, 1069–1075. [Google Scholar] [CrossRef]
- Chénard, C.; Wirth, J.F.; Suttle, C.A. Viruses infecting a freshwater filamentous cyanobacterium (Nostoc sp.) encode a functional CRISPR array and a proteobacterial DNA polymerase B. mBio 2016, 7, e00667-16. [Google Scholar] [CrossRef] [Green Version]
- Palermo, C.N.; Fulthorpe, R.R.; Saati, R.; Short, S.M. Metagenomic analysis of virus diversity and relative abundance in a eutrophic freshwater harbour. Viruses 2019, 11, 792. [Google Scholar] [CrossRef] [Green Version]
- Keshri, J.; Pradeep Ram, A.S.; Colombet, J.; Perriere, F.; Thouvenot, A.; Sime-Ngando, T. Differential impact of lytic viruses on the taxonomical resolution of freshwater bacterioplankton community structure. Water Res. 2017, 124, 129–138. [Google Scholar] [CrossRef]
- Rohwer, F.; Thurber, R.V. Viruses manipulate the marine environment. Nature 2009, 459, 207–212. [Google Scholar] [CrossRef]
- Culley, A.I. Insight into the unknown marine virus majority. Proc. Natl. Acad. Sci. USA 2013, 110, 12166–12167. [Google Scholar] [CrossRef] [Green Version]
- Lévesque, A.V.; Vincent, W.F.; Comte, J.; Lovejoy, C.; Culley, A.I. Chlorovirus and myovirus diversity in permafrost thaw ponds. Aquat. Microb. Ecol. 2018, 82, 209–224. [Google Scholar] [CrossRef]
- Shurin, J.B.; Clasen, J.L.; Greig, H.S.; Kratina, P.; Thompson, P.L. Warming shifts top-down and bottom-up control of pond food web structure and function. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 3008–3017. [Google Scholar] [CrossRef]
- Emerson, J.B.; Roux, S.; Brum, J.R.; Bolduc, B.; Woodcroft, B.J.; Jang, H.B.; Singleton, C.M.; Solden, L.M.; Naas, A.E.; Boyd, J.A.; et al. Host-linked soil viral ecology along a permafrost thaw gradient. Nat. Microbiol. 2018, 3, 870–880. [Google Scholar] [CrossRef]
- Vincent, W.F.; Lemay, M.; Allard, M. Arctic permafrost landscapes in transition: Towards an integrated Earth system approach. Arct. Sci. 2017, 3, 39–64. [Google Scholar] [CrossRef] [Green Version]
- Wauthy, M.; Rautio, M.; Christoffersen, K.S.; Forsström, L.; Laurion, I.; Mariash, H.L.; Peura, S.; Vincent, W.F. Increasing dominance of terrigenous organic matter in circumpolar freshwaters due to permafrost thaw: Increasing allochthony in arctic freshwaters. Limnol. Oceanogr. Lett. 2018, 3, 186–198. [Google Scholar] [CrossRef] [Green Version]
- Matveev, A.; Laurion, I.; Deshpande, B.N.; Vincent, W.F. Concentrations of dissolved methane, carbon dioxide and oxygen in thermokarst lakes and ponds in palsa peatlands, Northern Quebec, Canada, v. 1.100000 (2012–2017). Nordicana 2020, D48. [Google Scholar] [CrossRef]
- Cruaud, P.; Vigneron, A.; Fradette, M.-S.; Charette, S.J.; Rodriguez, M.J.; Dorea, C.C.; Culley, A.I. Open the SterivexTM casing: An easy and effective way to improve DNA extraction yields: DNA extraction from SterivexTM filters. Limnol. Oceanogr. Methods 2017, 15, 1015–1020. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 21 October 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinf. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Altschu, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Bolduc, B.; Youens-Clark, K.; Roux, S.; Hurwitz, B.L.; Sullivan, M.B. iVirus: Facilitating new insights in viral ecology with software and community data sets imbedded in a cyberinfrastructure. ISME J. 2017, 11, 7–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-R, L.M.; Gunturu, S.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. Nonpareil 3: Fast Estimation of metagenomic coverage and sequence diversity. MSystems 2018, 3, e00039-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; Version 3.6.1; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef] [Green Version]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Paez-Espino, D.; Pavlopoulos, G.A.; Ivanova, N.N.; Kyrpides, N.C. Nontargeted virus sequence discovery pipeline and virus clustering for metagenomic data. Nat. Protoc. 2017, 12, 1673–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A.; et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 2019, 37, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Pollard, K.S. Toward accurate and quantitative comparative metagenomics. Cell 2016, 166, 1103–1116. [Google Scholar] [CrossRef] [Green Version]
- Przytulska, A.; Comte, J.; Crevecoeur, S.; Lovejoy, C.; Laurion, I.; Vincent, W.F. Phototrophic pigment diversity and picophytoplankton in permafrost thaw lakes. Biogeosciences 2016, 13, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Brum, J.R.; Hurwitz, B.L.; Schofield, O.; Ducklow, H.W.; Sullivan, M.B. Seasonal time bombs: Dominant temperate viruses affect Southern Ocean microbial dynamics. ISME J. 2016, 10, 437–449. [Google Scholar] [CrossRef]
- Khot, V.; Strous, M.; Hawley, A.K. Computational approaches in viral ecology. Comput. Struct. Biotechnol. J. 2020, 18, 1605–1612. [Google Scholar] [CrossRef]
- Paez-Espino, D.; Chen, I.-M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Szeto, E.; Pillay, M.; Huang, J.; Markowitz, V.M.; Nielsen, T.; et al. IMG/VR: A database of cultured and uncultured DNA Viruses and retroviruses. Nucleic Acids Res. 2016, 45, gkw1030. [Google Scholar] [CrossRef]
- Breitbart, M.; Miyake, J.H.; Rohwer, F. Global distribution of nearly identical phage-encoded DNA sequences. FEMS Microbiol. Lett. 2004, 236, 249–256. [Google Scholar] [CrossRef]
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef]
- Roux, S.; Chan, L.-K.; Egan, R.; Malmstrom, R.R.; McMahon, K.D.; Sullivan, M.B. Ecogenomics of virophages and their giant virus hosts assessed through time series metagenomics. Nat. Commun. 2017, 8, 858. [Google Scholar] [CrossRef] [PubMed]
- Bellas, C.M.; Schroeder, D.C.; Edwards, A.; Barker, G.; Anesio, A.M. Flexible genes establish widespread bacteriophage pan-genomes in cryoconite hole ecosystems. Nat. Commun. 2020, 11, 4403. [Google Scholar] [CrossRef] [PubMed]
- Linz, A.M.; Crary, B.C.; Shade, A.; Owens, S.; Gilbert, J.A.; Knight, R.; McMahon, K.D. Bacterial community composition and dynamics spanning five years in freshwater bog lakes. mSphere 2017, 2, e00169-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharoni, S.; Trainic, M.; Schatz, D.; Lehahn, Y.; Flores, M.J.; Bidle, K.D.; Ben-Dor, S.; Rudich, Y.; Koren, I.; Vardi, A. Infection of phytoplankton by aerosolized marine viruses. Proc. Natl. Acad. Sci. USA 2015, 112, 6643–6647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reche, I.; D’Orta, G.; Mladenov, N.; Winget, D.M.; Suttle, C.A. Deposition rates of viruses and bacteria above the atmospheric boundary layer. ISME J. 2018, 12, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Gregory, A.C.; Zayed, A.A.; Conceição-Neto, N.; Temperton, B.; Bolduc, B.; Alberti, A.; Ardyna, M.; Arkhipova, K.; Carmichael, M.; Cruaud, C.; et al. Marine DNA viral macro- and microdiversity from pole to pole. Cell 2019, 177, 1109–1123. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Season | Cover (m) | Temperature (°C) | pH | Oxygen (mg L−1) | CH4 (µM) | SO4 (nM) | TN (mg L−1) | DOC (mg L−1) |
|---|---|---|---|---|---|---|---|---|
| Summer | none | Surface: 15 Bottom: 6 | 6 | Surface: 4.13 Bottom: <LD | Surface: 2.5 Bottom: 300 | 1.46 | 0.7 | 13.7 |
| Winter | 0.5 m snow 0.6 m ice | Surface: 0 Bottom: 3.5 | 5 | <LD | 200 | 0.5 | 1.2 | 18.3 |
| Genome Type | UViG | Contig ID | Length (bp) | Number of CDS | Summer Average Abundance | Winter Average Abundance | Putative Host Rank | Putative Viral Group |
|---|---|---|---|---|---|---|---|---|
| C | UViG3 | Ga0256681_10553986 | 63,992 | 103 | 2.18 | 0 | Firmicutes | Siphoviridae, Myoviridae (Lactococcus, Clostridium, Geobacillus phages) |
| C | UViG4 | Ga0256681_10542696 | 34,037 | 36 | 7.61 | 0 | ||
| C | UViG5 | Ga0256681_10559173 | 36,339 | 61 | 13.3 | 0 | Gammaproteobacteria | Podoviridae (Rhodoferax, Vibrio, Thalassomonas phages) |
| C | UViG6 | Ga0256681_11263128 | 62,403 | 87 | 4.54 | 0 | Gammaproteobacteria | Podoviridae, Sophoviridae (Pseudomonas, Xanthomonas phages) |
| C | UViG7 | Ga0256681_10579994 | 45,748 | 55 | 1.65 | 0 | ||
| C | UViG8 | Ga0256681_10547172 | 42,853 | 61 | 8.45 | 0 | Gammaproteobacteria | Podoviridae (Acetinobacter, Aeromonas, Edwardsiella phages) |
| C | UViG9 | Ga0256681_10567549 | 36,908 | 45 | 6.1 | 0 | Gammaproteobacteria | Podoviridae, Siphoviridae (Pseudoalteromonas, Marinomonas, Escherichia viruses) |
| C | UViG10 | Ga0256681_10572110 | 37,103 | 56 | 1.63 | 0 | Alphaproteobacteria | Siphoviridae (Caulobacter viruses) |
| C | UViG11 | Ga0256681_10539383 | 83,527 | 107 | 5.85 | 2.16 | Unknown | Unknown (Natrinema, Haloarcula viruses) |
| P | vOTU12 | Ga0256681_11878260 | 12,576 | 13 | 0.37 | 0 | Gammaproteobacteria | Myoviridae (Pseudomonas, Stenotrophomonas, Xanthomonas phage) |
| P | vOTU13 | Ga0256681_12578680 | 29,945 | 13 | 9.71 | 0 | (Bacteria, Archaea) | Unknown |
| P | vOTU14 | Ga0256681_12584168 | 15,432 | 17 | 1.2 | 0 | Unknown | Unknown |
| P | vOTU15 | Ga0256681_12612509 | 13,421 | 11 | 2.46 | 0 | (Bacteria) | Unknown |
| P | vOTU16 | Ga0256681_10168136 | 15,698 | 15 | 0.89 | 0 | Betaproteobacteria (Polaromonas) | Unknown |
| P | vOTU17 | Ga0256681_10683670 | 11,399 | 18 | 3.47 | 0 | Unknown | Unknown |
| P | vOTU18 | Ga0256681_11428677 | 14,176 | 18 | 1.33 | 0 | (Bacteria, Archaea) | Unknown |
| P | vOTU21 | Ga0256681_10088446 | 10,785 | 11 | 9.7 | 0 | (Bacteria, Archaea) | Unknown |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girard, C.; Langlois, V.; Vigneron, A.; Vincent, W.F.; Culley, A.I. Seasonal Regime Shift in the Viral Communities of a Permafrost Thaw Lake. Viruses 2020, 12, 1204. https://doi.org/10.3390/v12111204
Girard C, Langlois V, Vigneron A, Vincent WF, Culley AI. Seasonal Regime Shift in the Viral Communities of a Permafrost Thaw Lake. Viruses. 2020; 12(11):1204. https://doi.org/10.3390/v12111204
Chicago/Turabian StyleGirard, Catherine, Valérie Langlois, Adrien Vigneron, Warwick F. Vincent, and Alexander I. Culley. 2020. "Seasonal Regime Shift in the Viral Communities of a Permafrost Thaw Lake" Viruses 12, no. 11: 1204. https://doi.org/10.3390/v12111204
APA StyleGirard, C., Langlois, V., Vigneron, A., Vincent, W. F., & Culley, A. I. (2020). Seasonal Regime Shift in the Viral Communities of a Permafrost Thaw Lake. Viruses, 12(11), 1204. https://doi.org/10.3390/v12111204