The Structure of an AAV5-AAVR Complex at 2.5 Å Resolution: Implications for Cellular Entry and Immune Neutralization of AAV Gene Therapy Vectors

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Expression of AAV5 and AAVR
2.2. Single Particle Cryo-EM
2.3. Atomic Modeling and Refinement
2.4. Feasibility of Two-Domain Binding
2.5. ELISA Binding Assays
3. Results and Discussion
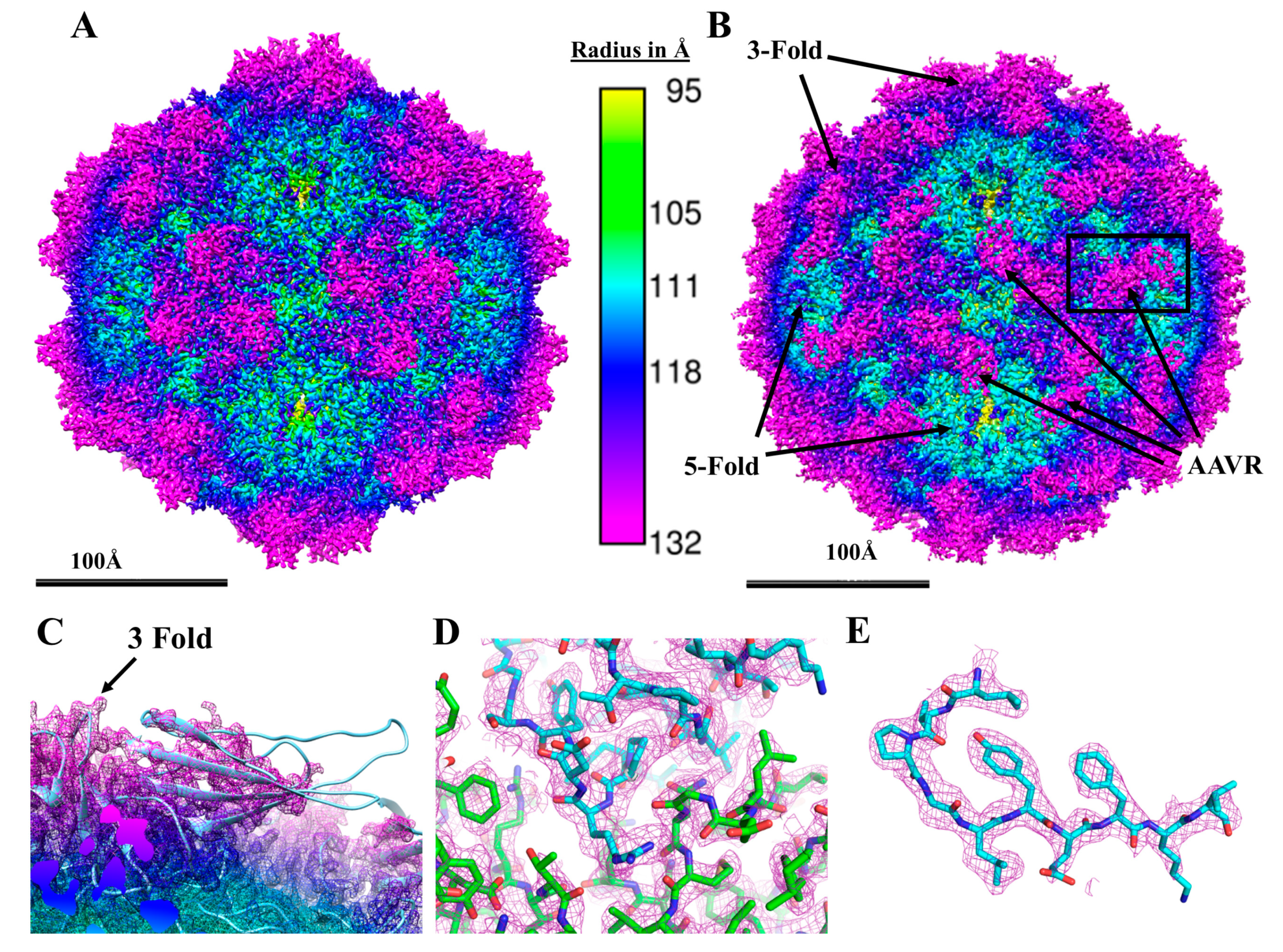
3.1. High-Resolution Structure of Native AAV5 and Bound AAV5-PKD12
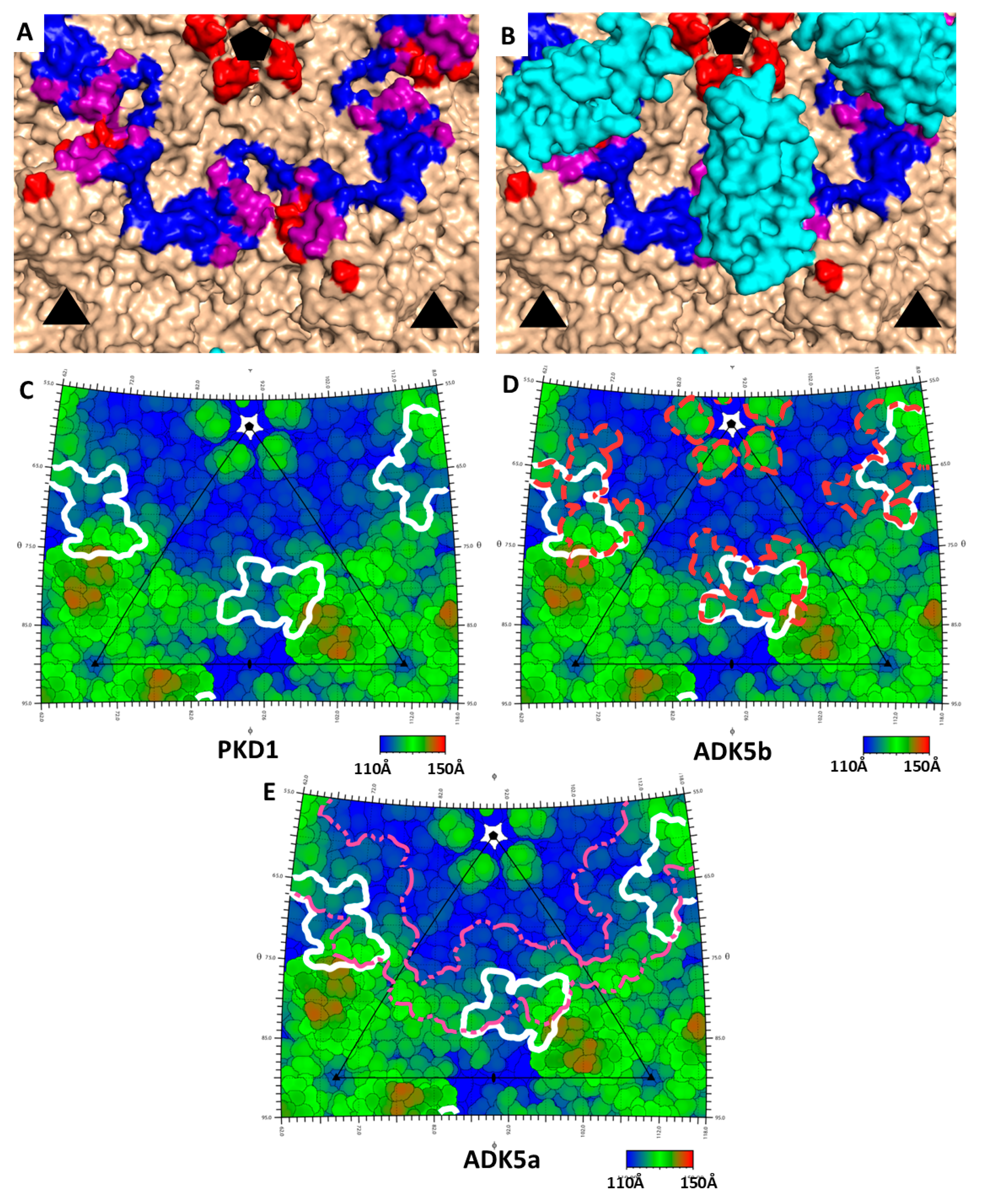
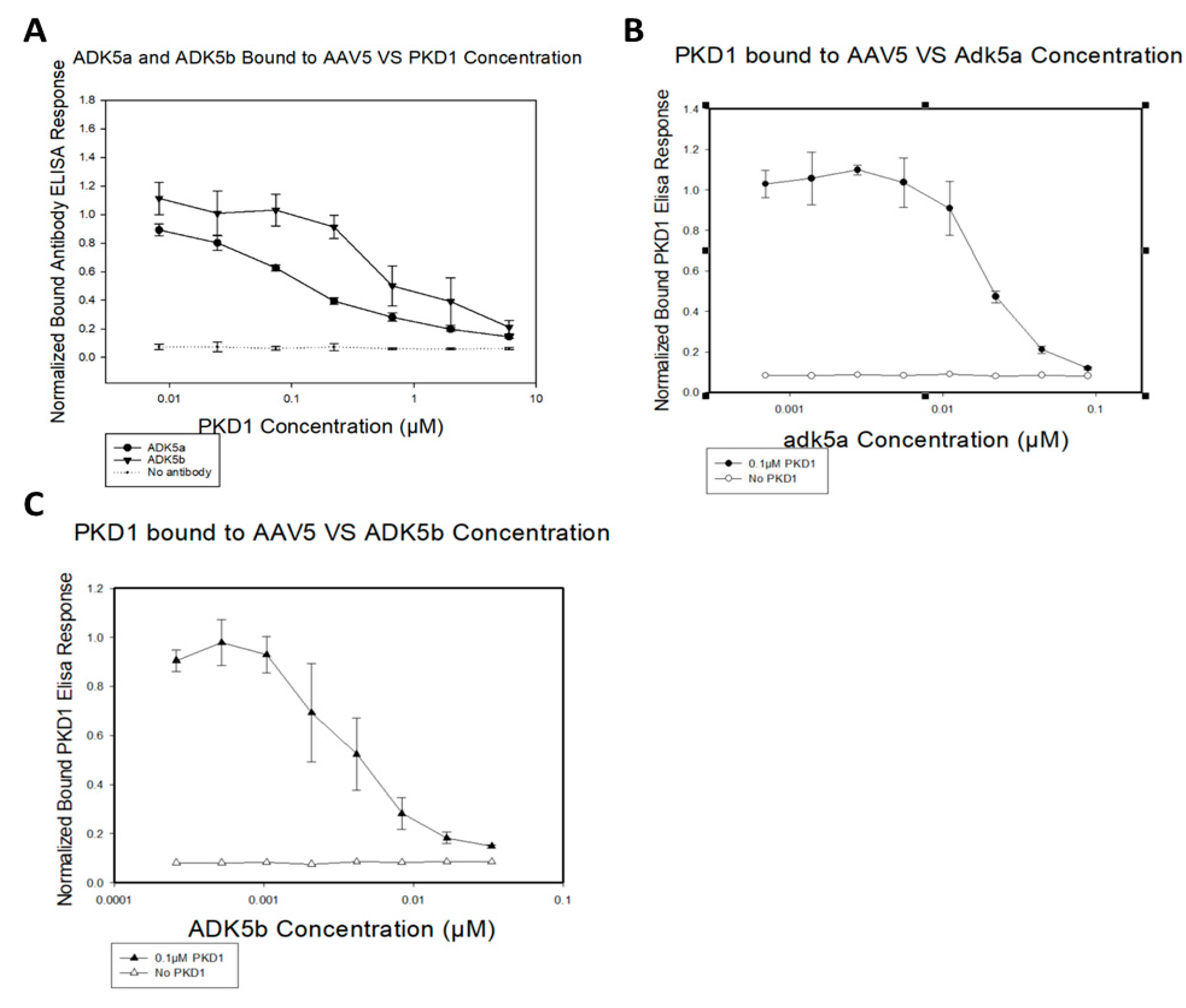
3.2. Antibody Neutralization of AAV5 and AAVR
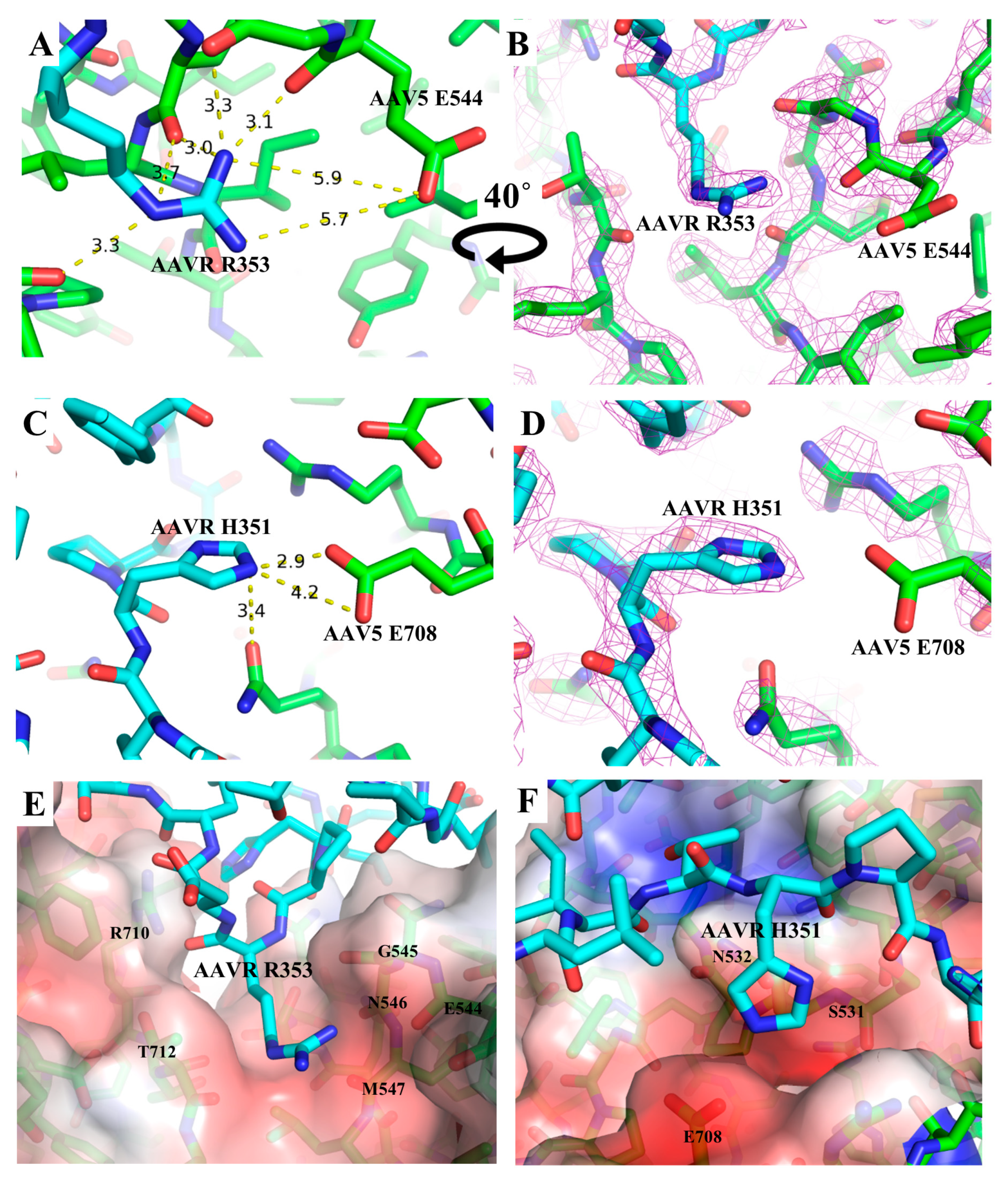
3.3. PKD1 Binding Site on AAV2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tattersall, P. The Evolution of Parvovirus Taxonomy. In Parvoviruses; Bloom, M.E., Cotmore, S.F., Linden, R.M., Parrish, C.R., Kerr, J.R., Eds.; Hodder Arnold: London, UK, 2006; pp. 5–14. [Google Scholar]
- Atchison, R.W.; Casto, B.C.; Hammon, W.M. Adenovirus-Associated Defective Virus Particles. Science 1965, 149, 754–755. [Google Scholar] [CrossRef]
- Conway, J.E.; Zolotukhin, S.; Muzyczka, N.; Hayward, G.S.; Byrne, B.J. Recombinant adeno-associated virus type 2 replication and packaging is entirely supported by a herpes simplex virus type 1 amplicon expressing Rep and Cap. J. Virol. 1997, 71, 8780–8789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, A.F.; Fraefel, C.; Seyffert, M. The Interplay between Adeno-Associated Virus and Its Helper Viruses. Viruses 2020, 12, 662. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Lusby, E.W.; Berns, K.I. Nucleotide sequence and organization of the adeno-associated virus 2 genome. J. Virol. 1983, 45, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [Green Version]
- Keeler, A.M.; Flotte, T.R. Recombinant Adeno-Associated Virus Gene Therapy in Light of Luxturna (and Zolgensma and Glybera): Where Are We, and How Did We Get Here? Annu. Rev. Virol. 2019, 6, 601–621. [Google Scholar] [CrossRef]
- Smalley, E. First AAV gene therapy poised for landmark approval. Nat. Biotechnol. 2017, 35, 998–999. [Google Scholar] [CrossRef]
- Al-Zaidy, S.; Pickard, A.S.; Kotha, K.; Alfano, L.N.; Lowes, L.; Paul, G.; Church, K.; Lehman, K.; Sproule, D.M.; Dabbous, O.; et al. Health outcomes in spinal muscular atrophy type 1 following AVXS-101 gene replacement therapy. Pediatr. Pulmonol. 2019, 54, 179–185. [Google Scholar] [CrossRef]
- Srivastava, A. In vivo tissue-tropism of adeno-associated viral vectors. Curr. Opin. Virol. 2016, 21, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Hauck, B.; Xiao, W. Characterization of Tissue Tropism Determinants of Adeno-Associated Virus Type 1. J. Virol. 2003, 77, 2768–2774. [Google Scholar] [CrossRef] [Green Version]
- Dudek, A.M.; Pillay, S.; Puschnik, A.S.; Nagamine, C.M.; Cheng, F.; Qiu, J.; Carette, J.E.; Vandenberghe, L.H. An Alternate Route for Adeno-associated Virus (AAV) Entry Independent of AAV Receptor. J. Virol. 2018, 92, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Pillay, S.; Meyer, N.L.; Puschnik, A.S.; Davulcu, O.; Diep, J.; Ishikawa, Y.; Jae, L.T.; Wosen, J.E.; Nagamine, C.M.; Chapman, M.S.; et al. An essential receptor for adeno-associated virus infection. Nature 2016, 530, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, B.A.; Li, X.; Pezzulo, A.A.; Alaiwa, M.H.A.; Zabner, J. Polarized AAVR Expression Determines Infectivity by AAV Gene Therapy Vectors. Physiol. Behav. 2019, 26, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Pillay, S.; Zou, W.; Cheng, F.; Puschnik, A.S.; Meyer, N.L.; Ganaie, S.S.; Deng, X.; Wosen, J.E.; Davulcu, O.; Yan, Z.; et al. Adeno-associated Virus (AAV) Serotypes Have Distinctive Interactions with Domains of the Cellular AAV Receptor. J. Virol. 2017, 91, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Xu, G.; Cao, L.; Sun, Z.; He, Y.; Cui, M.; Sun, Y.; Li, S.; Li, H.; Qin, L.; et al. Divergent engagements between adeno-associated viruses with their cellular receptor AAVR. Nat. Commun. 2019, 10, 3760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Cao, L.; Cui, M.; Sun, Z.; Hu, M.; Zhang, R.; Stuart, W.; Zhao, X.; Yang, Z.; Li, X.; et al. Adeno-associated virus 2 bound to its cellular receptor AAVR. Nat. Microbiol. 2019, 4, 675–682. [Google Scholar] [CrossRef]
- Meyer, N.L.; Hu, G.; Davulcu, O.; Xie, Q.; Noble, A.J.; Yoshioka, C.; Gingerich, D.S.; Trzynka, A.; David, L.; Stagg, S.M.; et al. Structure of the gene therapy vector, adeno-associated virus with its cell receptor, AAVR. Elife 2019, 8, e44707. [Google Scholar] [CrossRef]
- Urabe, M.; Ding, C.; Kotin, R.M. Insect Cells as a Factory to Produce Adeno-Associated Virus Type 2 Vectors. Hum. Gene Ther. 2002, 13, 1935–1943. [Google Scholar] [CrossRef]
- Meyer, N.; Davulcu, O.; Xie, Q.; Silveria, M.; Zane, G.M.; Large, E.; Chapman, M.S. Expression and Purification of Adeno-associated Virus Virus-like Particles in a Baculovirus System and AAVR Ectodomain Constructs in E. coli Nancy. Bio-Protocol 2020, 10. [Google Scholar] [CrossRef]
- Zivanov, J.; Nakane, T.; Forsberg, B.O.; Kimanius, D.; Hagen, W.J.H.; Lindahl, E.; Scheres, S.H.W. New tools for automated high-resolution cryo-EM structure determination in RELION-3. ELife 2018, 7, e42166. [Google Scholar] [CrossRef]
- Rohou, A.; Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015, 192, 216–221. [Google Scholar] [CrossRef]
- Govindasamy, L.; DiMattia, M.A.; Gurda, B.L.; Halder, S.; McKenna, R.; Chiorini, J.A.; Muzyczka, N.; Zolotukhin, S.; Agbandje-McKenna, M. Structural Insights into Adeno-Associated Virus Serotype 5. J. Virol. 2013, 87, 11187–11199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, M.S. Restrained real-space macromolecular atomic refinement using a new resolution-dependent electron-density function. Acta Crystallogr. Sect. A 1995, 51, 69–80. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinforma. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Brünger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography and NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 905–921. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Govindasamy, L.; Padron, E.; McKenna, R.; Muzyczka, N.; Kaludov, N.; Chiorini, J.A.; Agbandje-McKenna, M. Structurally Mapping the Diverse Phenotype of Adeno-Associated Virus Serotype 4. J. Virol. 2006, 80, 11556–11570. [Google Scholar] [CrossRef] [Green Version]
- Bantel-Schaal, U.; Delius, H.; Schmidt, R.; zur Hausen, H. Human adeno-associated virus type 5 is only distantly related to other known primate helper-dependent parvoviruses. J. Virol. 1999, 73, 939–947. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Bartesaghi, A.; Matthies, D.; Banerjee, S.; Merk, A.; Subramaniam, S. Structure of β-galactosidase at 3.2-Å resolution obtained by cryo-electron microscopy. Proc. Natl. Acad. Sci. USA 2014, 111, 11709–11714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, N.A. Poisson–Boltzmann Methods for Biomolecular Electrostatics. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2004; Volume 383, pp. 94–118. [Google Scholar]
- Ilca, S.L.; Kotecha, A.; Sun, X.; Poranen, M.M.; Stuart, D.I.; Huiskonen, J.T. Localized reconstruction of subunits from electron cryomicroscopy images of macromolecular complexes. Nat. Commun. 2015, 6, 8843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, Y.-S.; Gurda, B.L.; Chipman, P.; McKenna, R.; Afione, S.; Chiorini, J.A.; Muzyczka, N.; Olson, N.H.; Baker, T.S.; Kleinschmidt, J.; et al. Adeno-Associated Virus Serotype 1 (AAV1)- and AAV5-Antibody Complex Structures Reveal Evolutionary Commonalities in Parvovirus Antigenic Reactivity. J. Virol. 2015, 89, 1794–1808. [Google Scholar] [CrossRef] [Green Version]
- Jose, A.; Mietzsch, M.; Smith, J.K.; Kurian, J.; Chipman, P.; McKenna, R.; Chiorini, J.; Agbandje-McKenna, M. High-Resolution Structural Characterization of a New Adeno-associated Virus Serotype 5 Antibody Epitope toward Engineering Antibody-Resistant Recombinant Gene Delivery Vectors. J. Virol. 2018, 93, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Gurda, B.L.; DiMattia, M.A.; Miller, E.B.; Bennett, A.; McKenna, R.; Weichert, W.S.; Nelson, C.D.; Chen, W.-J.; Muzyczka, N.; Olson, N.H.; et al. Agbandje-McKenna, M. Capsid Antibodies to Different Adeno-Associated Virus Serotypes Bind Common Regions. J. Virol. 2013, 87, 9111–9124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, M.S. Mapping the surface properties of macromolecules. Protein Sci. 1993, 2, 459–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, C.; Rossmann, M.G. Interpretation of electron density with stereographic roadmap projections. J Struct. Biol. 2007, 158, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Harbison, C.E.; Weichert, W.S.; Gurda, B.L.; Chiorini, J.A.; Agbandje-McKenna, M.; Parrish, C.R. Examining the cross-reactivity and neutralization mechanisms of a panel of mAbs against adeno-associated virus serotypes 1 and 5. J. Gen. Virol. 2012, 93, 347–355. [Google Scholar] [CrossRef]
- Afione, S.; DiMattia, M.A.; Halder, S.; Di Pasquale, G.; Agbandje-McKenna, M.; Chiorini, J.A. Identification and Mutagenesis of the Adeno-Associated Virus 5 Sialic Acid Binding Region. J. Virol. 2015, 89, 1660–1672. [Google Scholar] [CrossRef] [Green Version]
- Januliene, D.; Manavalan, A.; Ovesen, P.L.; Pedersen, K.-M.; Thirup, S.; Nykjær, A.; Moeller, A. Hidden Twins: SorCS Neuroreceptors Form Stable Dimers. J. Mol. Biol. 2017, 429, 2907–2917. [Google Scholar] [CrossRef]
- Leloup, N.; Chataigner, L.M.P.; Janssen, B.J.C. Structural insights into SorCS2–Nerve Growth Factor complex formation. Nat. Commun. 2018, 9, 2979. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
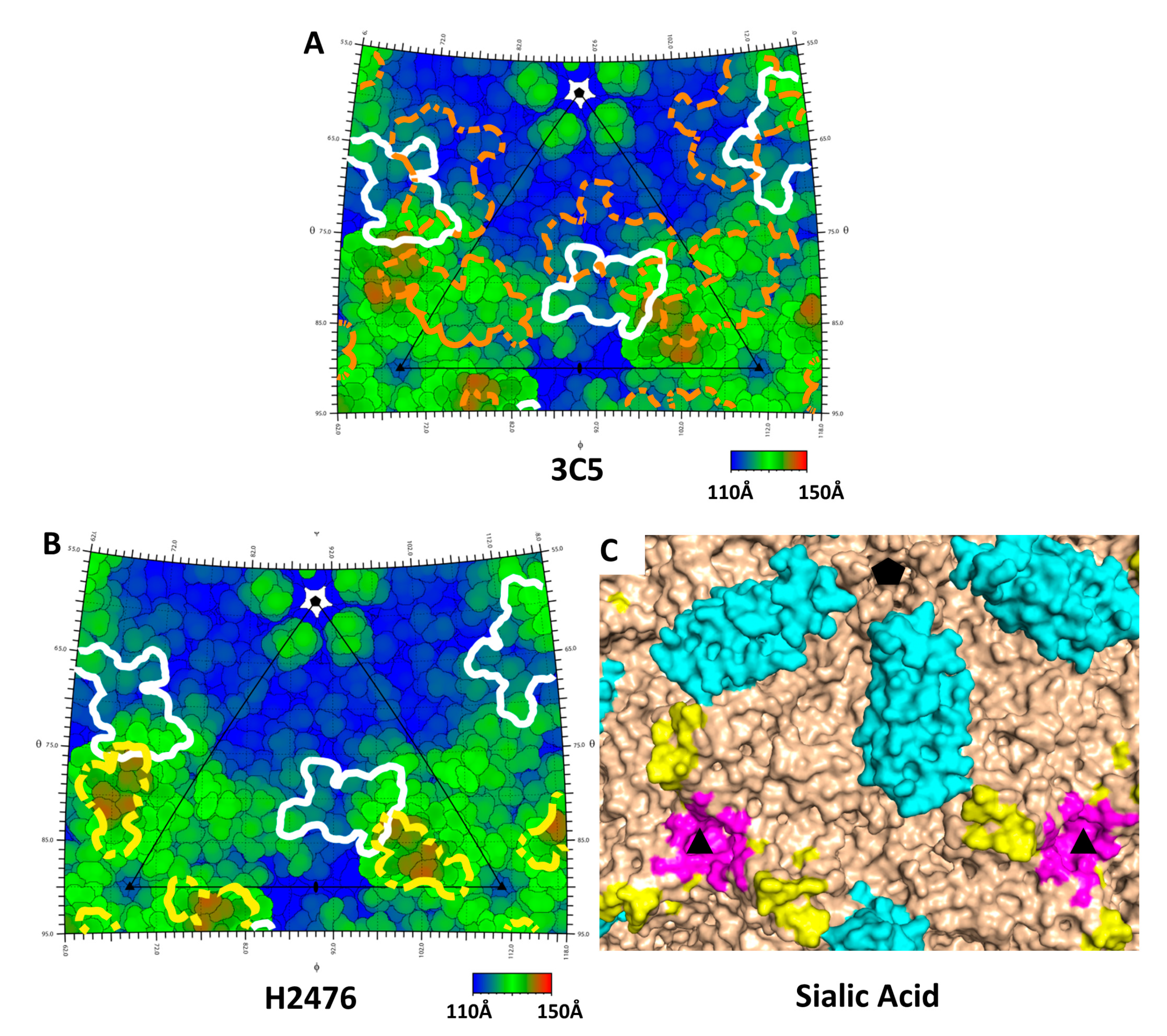{kind=link}
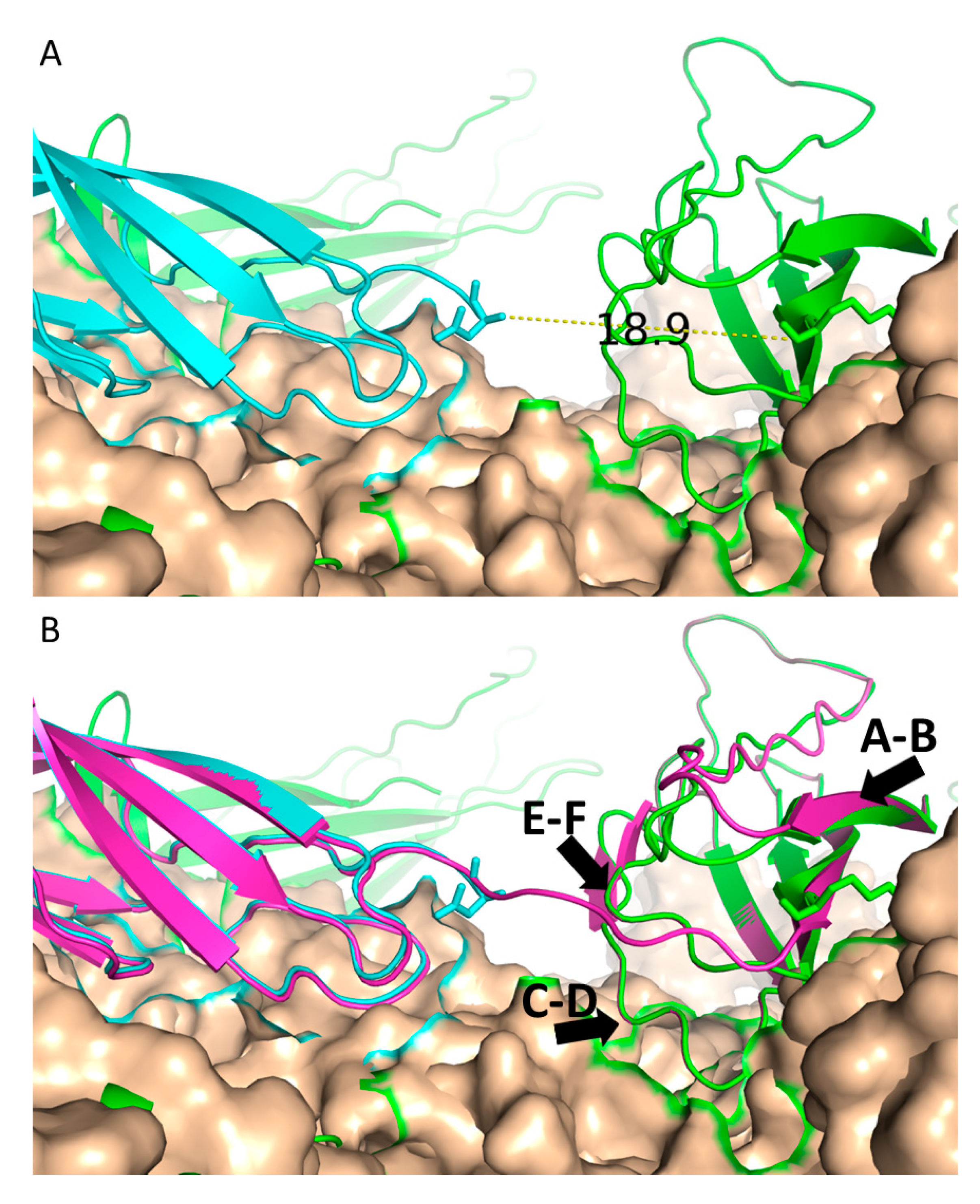{kind=link}
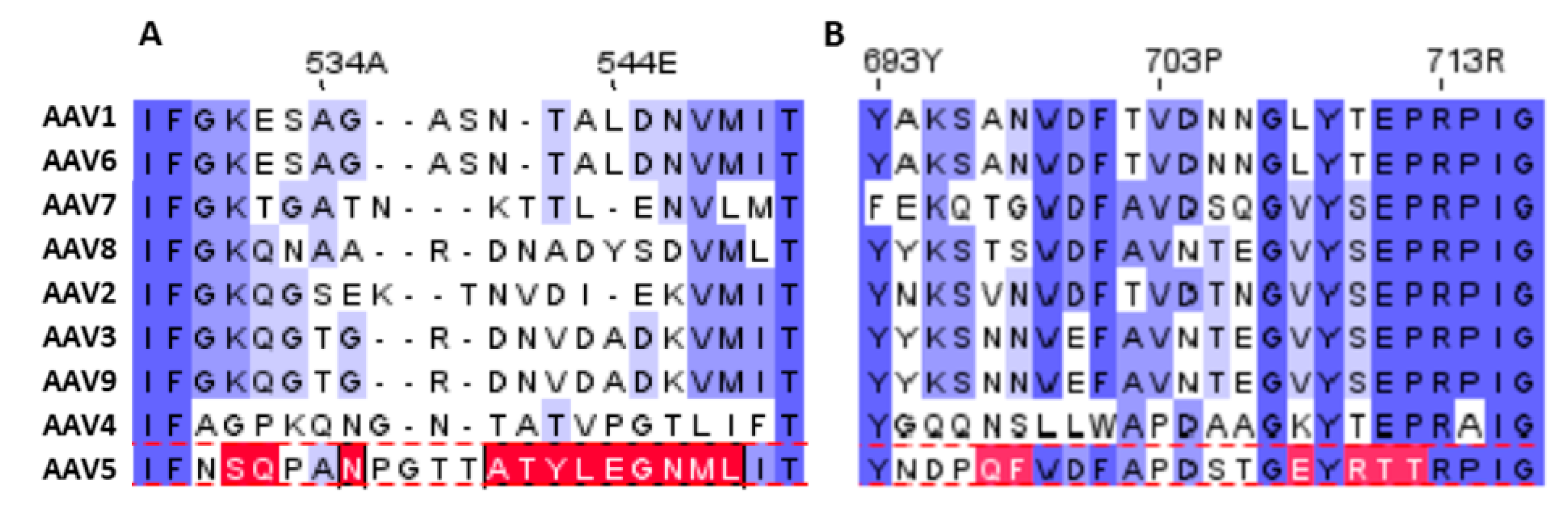{kind=link}
| Data Collection | AAV5 (Uncomplexed) | AAVR-AAV5 Complex |
|---|---|---|
| Magnification | 81,000× | 64,000× |
| Voltage | 300 kV | 300 kV |
| Electron exposure | 31.7 e−/Å2 | 32.9e−/Å2 |
| Defocus range | −0.7 to −2 μm | −0.7 to −2.5 μm |
| Pixel size | 0.530 Å | 0.664 Å |
| (on refinement vs. atomic model:) | 0.533 Å | 0.665 Å |
| Data processing | ||
| Motion correction | Relion 3.1-beta | Relion 3.0 |
| CTF estimation | CTFFIND-4.1 | CTFFIND-4.1 |
| Symmetry imposed | I1 | I1 |
| Initial particle images | 672,106 | 168,275 |
| Final particle images | 373,426 | 159,673 |
| Map resolution | 2.13 Å | 2.5 Å |
| FSC threshold | 0.143 | 0.143 |
| AAV5 (Uncomplexed) | AAVR-AAV5 Complex | |
|---|---|---|
| Protein atoms/asymmetric unit (mean B-factor): | 4096 (18.6 Å2) | 4794 (28.83 Å2) |
| RMS bond length deviation from ideal | 0.011 Å | 0.016 Å |
| RMS bond angle deviation from ideal | 1.4° | 1.8° |
| Ramachandran outliers | 2 (0.4%) | 4 (1%) |
| Cross-correlation (model-map) | 0.833 | 0.883 |
| Resolution from model-map refinement (d0.5) | 1.91 Å | 2.51 Å |
| Atoms from AAVR-AAV5 Complex at 2.5 Å Resolution (This Paper) | Alignment | RMSD vs. AAVR-AAV5 Complex at 3.2 Å Resolution [16] | RMSD vs. AAV5 at 2.1 Å Resolution (Uncomplexed, This Paper) |
|---|---|---|---|
| All protein atoms | By point-group symmetry | 1.3 Å | 0.44 Å |
| All protein atoms | All-atom least-squares | 0.94 Å | 0.43 Å |
| AAV5, all atoms | All-atom least-squares | 0.8 Å | 0.4 Å |
| AAV5, backbone | All-atom least-squares | 0.5 Å | 0.3 Å |
| AAV5, side chains | All-atom least-squares | 1.0 Å | 0.5 Å |
| AAVR (PKD1), all atoms | All-atom least-squares | 2.3 Å | n/a |
| AAVR (PKD1), backbone | All-atom least-squares | 1.9 Å | n/a |
| AAVR (PKD1), side chains | All-atom least-squares | 2.6 Å | n/a |
| AAVR | AAV5 | Donor to Acceptor Distance (Å) |
|---|---|---|
| Ile349CO | Gln532Nε | 3.1 |
| His351Nε | Glu532Oε | 3.4 |
| Arg353Nε | Thr712CO | 3.3 |
| Arg353Nη | Gly545CO | 3.3 |
| Arg353Nη | Glu544CO | 3.1 |
| Arg353NH | Gly545CO | 3.2 |
| Arg353NH | Met547CO | 3.0 |
| Arg353CO | Arg710Nε | 3.2 |
| Tyr355CO | Arg710Nε | 2.4 |
| Lys371Nζ | Gln697Oε | 2.3 |
| Leu376NH | Asn546Oδ | 2.9 |
| Leu376CO | Asn546Nδ | 3.1 |
| AAVR Residues Involved in Hydrophobic Contacts with AAV5 | ||||||||||
| Thr350 | Pro352 | Asp354 | Ser356 | Pro374 | Gly375 | Leu376 | Thr397 | Lys399 | ||
| AAV5 Residues Involved in Hydrophobic Contacts with AAVR | ||||||||||
| Asn442 | Ser531 | Ala540 | Thr541 | Tyr542 | Leu543 | Asn546 | Met547 | Leu548 | Gln697 | Phe698 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silveria, M.A.; Large, E.E.; Zane, G.M.; White, T.A.; Chapman, M.S. The Structure of an AAV5-AAVR Complex at 2.5 Å Resolution: Implications for Cellular Entry and Immune Neutralization of AAV Gene Therapy Vectors. Viruses 2020, 12, 1326. https://doi.org/10.3390/v12111326
Silveria MA, Large EE, Zane GM, White TA, Chapman MS. The Structure of an AAV5-AAVR Complex at 2.5 Å Resolution: Implications for Cellular Entry and Immune Neutralization of AAV Gene Therapy Vectors. Viruses. 2020; 12(11):1326. https://doi.org/10.3390/v12111326
Chicago/Turabian StyleSilveria, Mark A., Edward E. Large, Grant M. Zane, Tommi A. White, and Michael S. Chapman. 2020. "The Structure of an AAV5-AAVR Complex at 2.5 Å Resolution: Implications for Cellular Entry and Immune Neutralization of AAV Gene Therapy Vectors" Viruses 12, no. 11: 1326. https://doi.org/10.3390/v12111326
APA StyleSilveria, M. A., Large, E. E., Zane, G. M., White, T. A., & Chapman, M. S. (2020). The Structure of an AAV5-AAVR Complex at 2.5 Å Resolution: Implications for Cellular Entry and Immune Neutralization of AAV Gene Therapy Vectors. Viruses, 12(11), 1326. https://doi.org/10.3390/v12111326