Extracellular Vesicle Activation of Latent HIV-1 Is Driven by EV-Associated c-Src and Cellular SRC-1 via the PI3K/AKT/mTOR Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Infection and Treatment of PBMCs
2.3. EV Isolation and Ultracentrifugation
2.4. EV Characterization Using ZetaView
2.5. Nanoparticle Capture of EVs/Virions
2.6. Cell Transfection
2.7. Cell Lysis
2.8. Western Blot Analysis
2.9. Cytotoxicity Assay
2.10. RNA Isolation and RT-qPCR
2.11. Kinase Assay
2.12. ChIP Assay
2.13. Statistical Analysis
3. Results
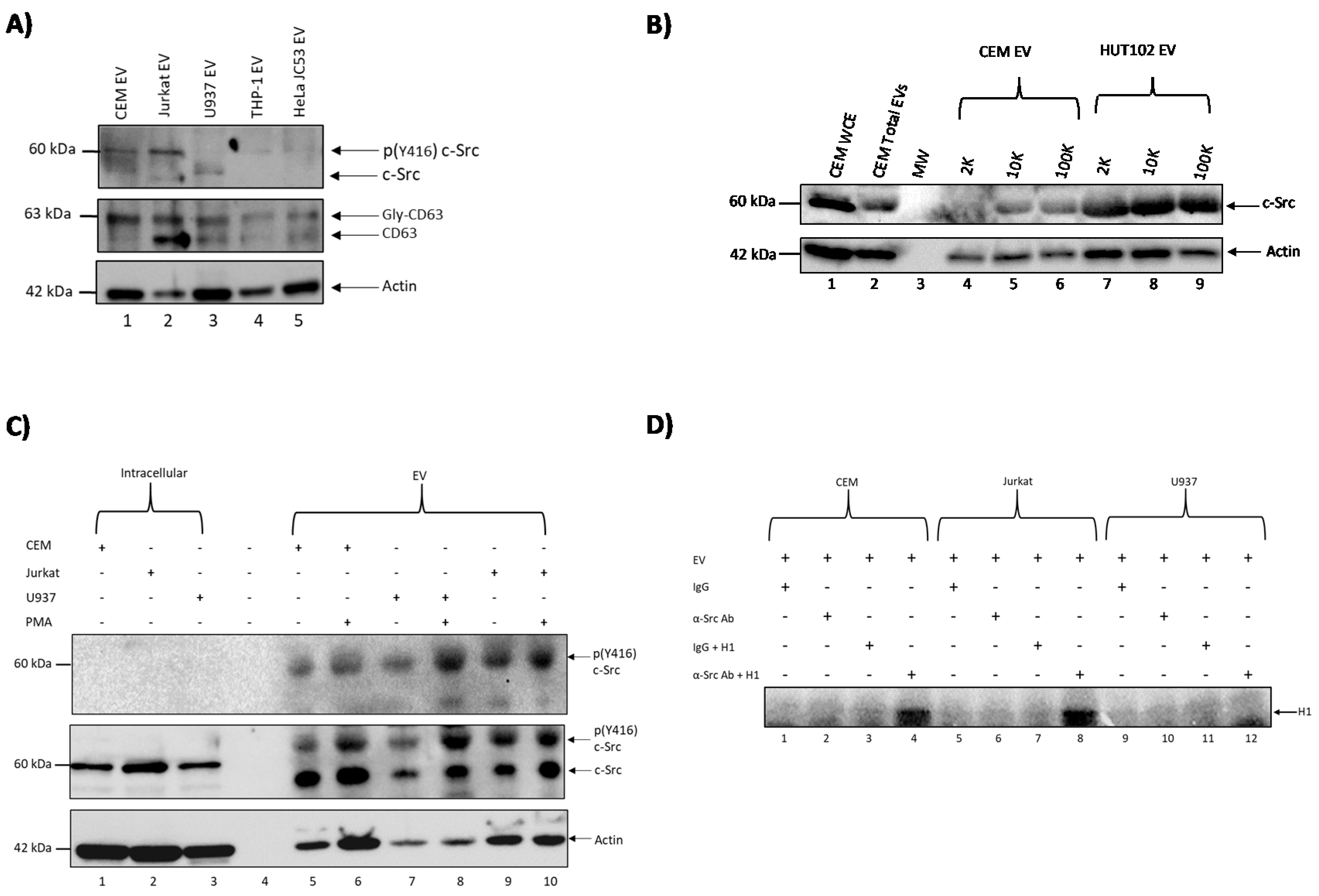
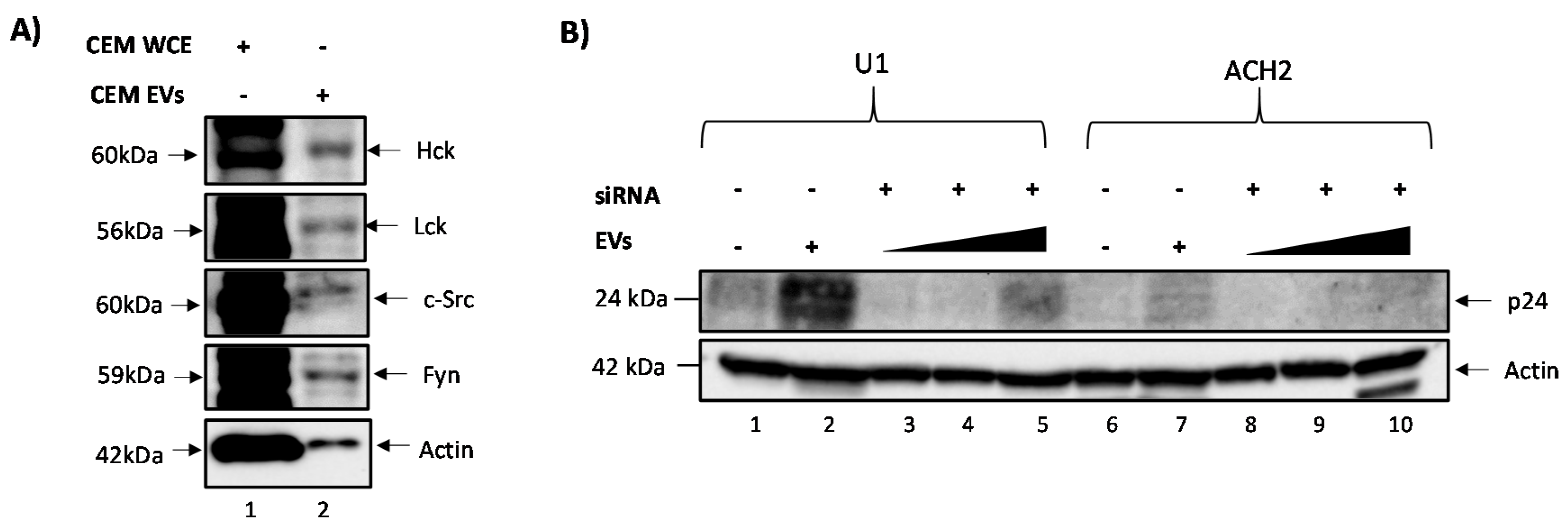
3.1. c-Src Is Present in Multiple Cell Lines and Different EV Populations
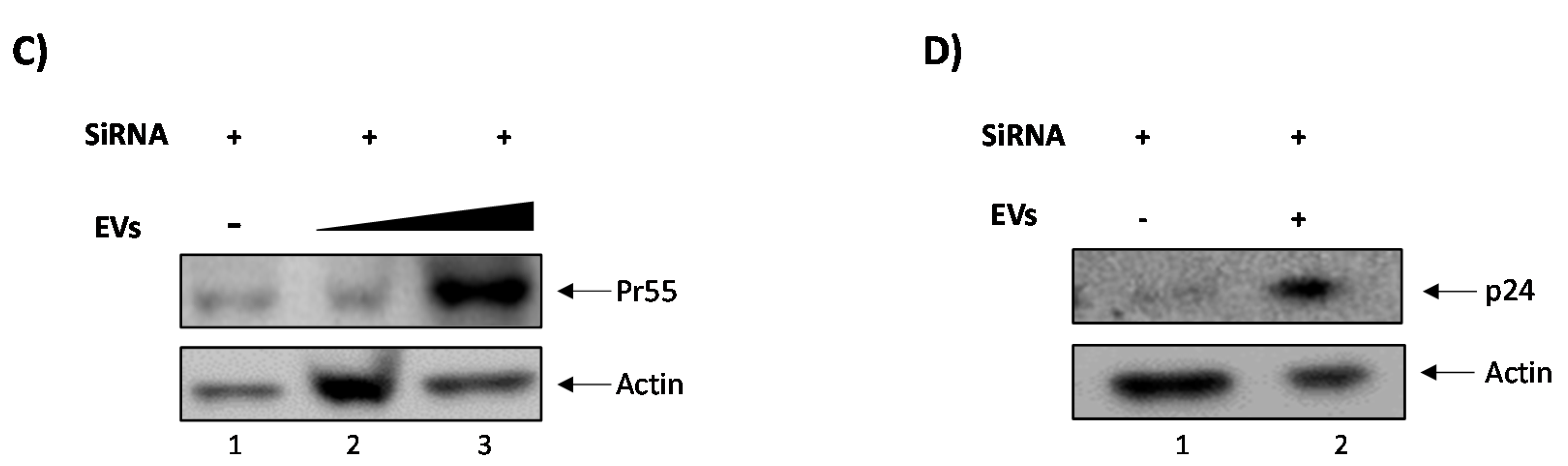
3.2. Elucidating the Activation Pathway of Latent HIV-1 by c-Src
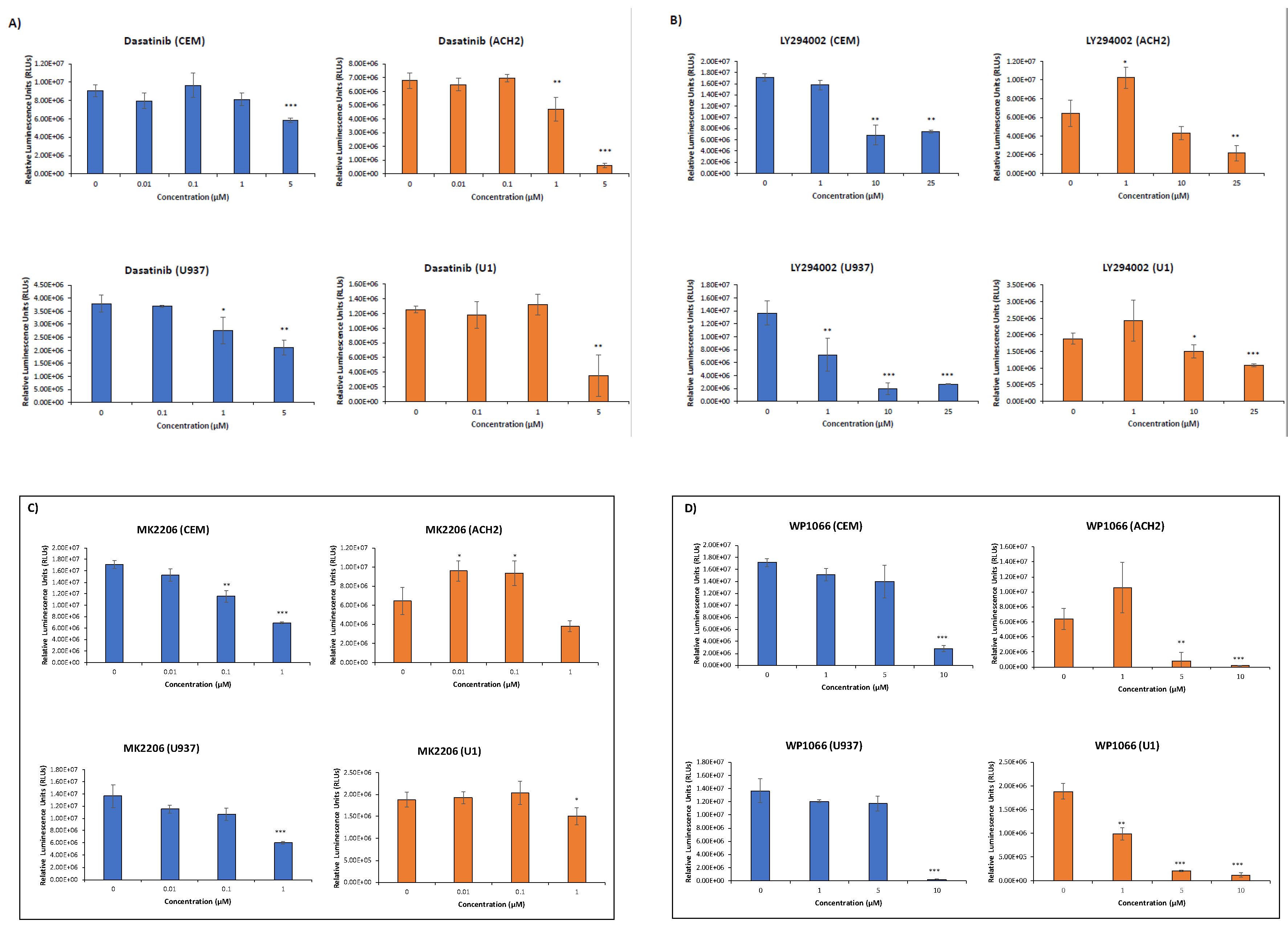
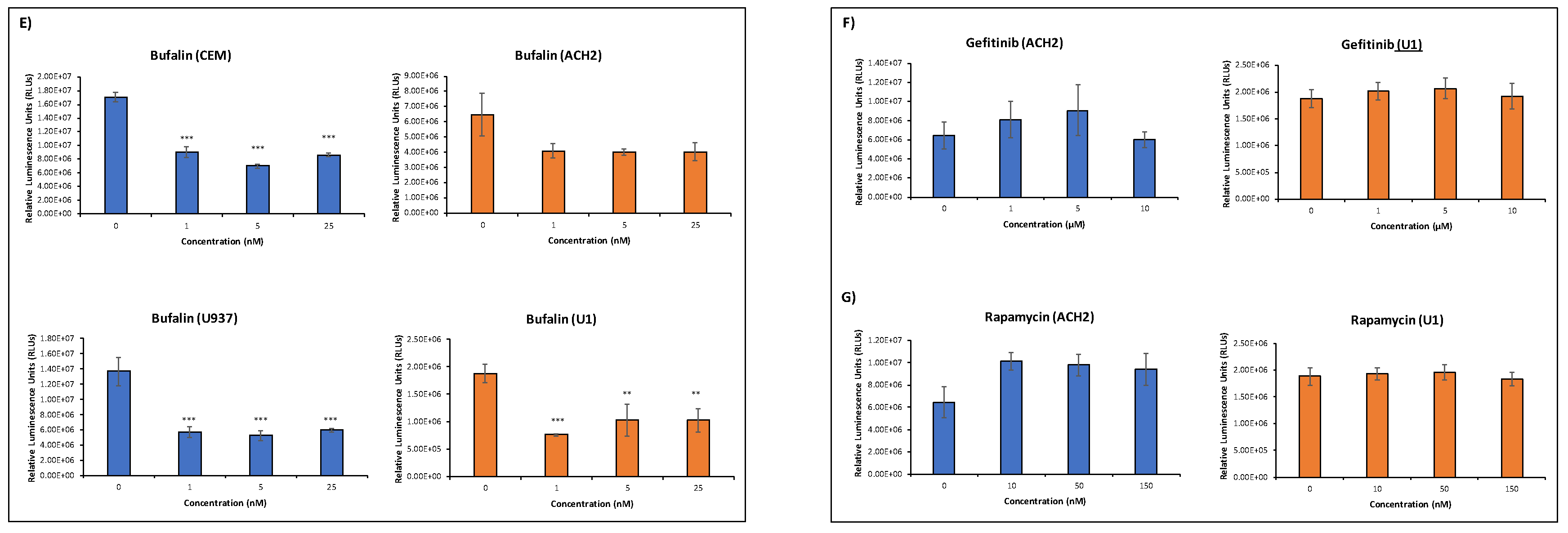
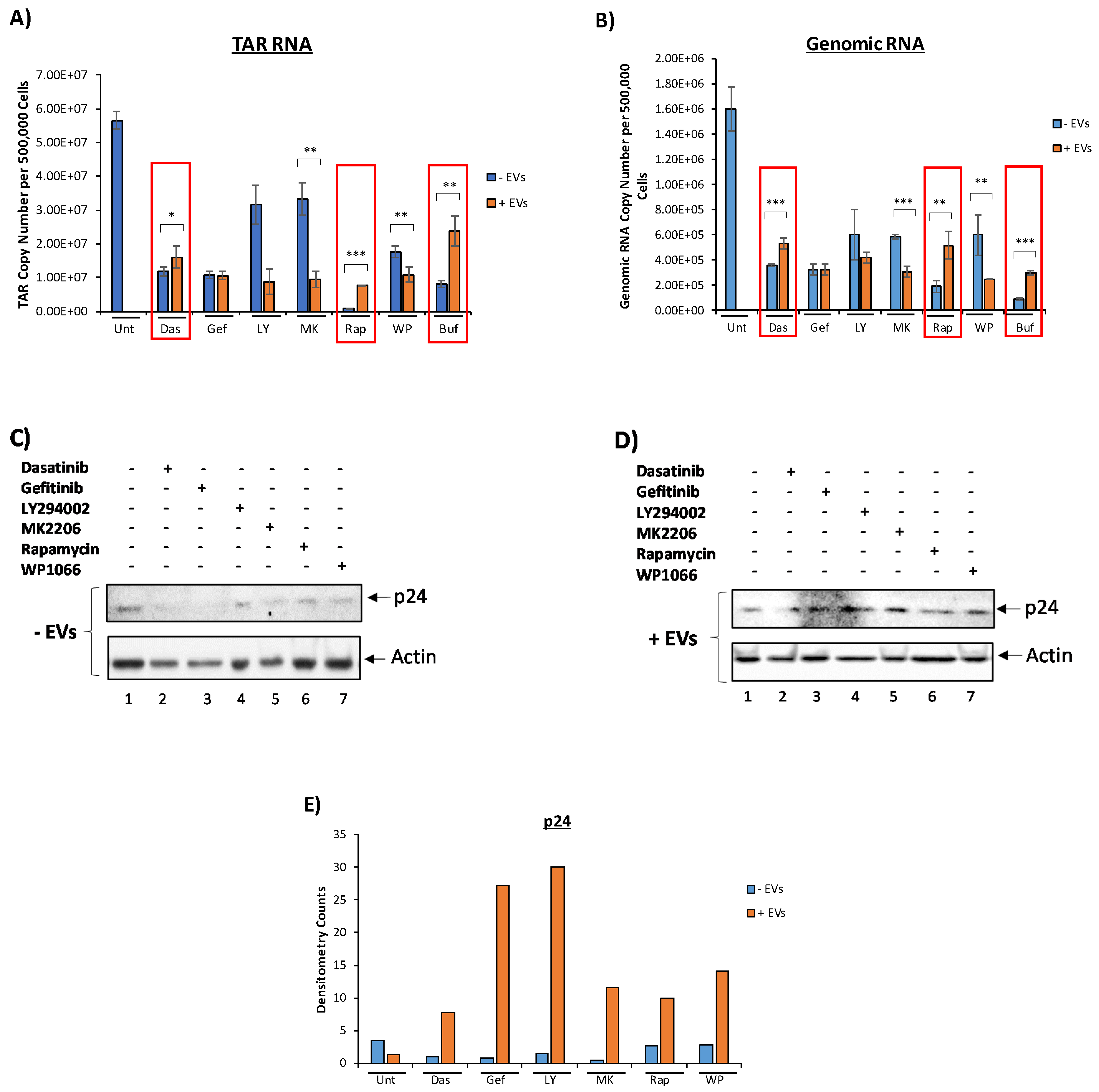
3.3. EVs Containing c-Src rescue HIV-1 Levels in Inhibitor-Treated Cells
3.4. Confirming EV-Associated c-Src Activates Latent HIV-1 in Infected Cells
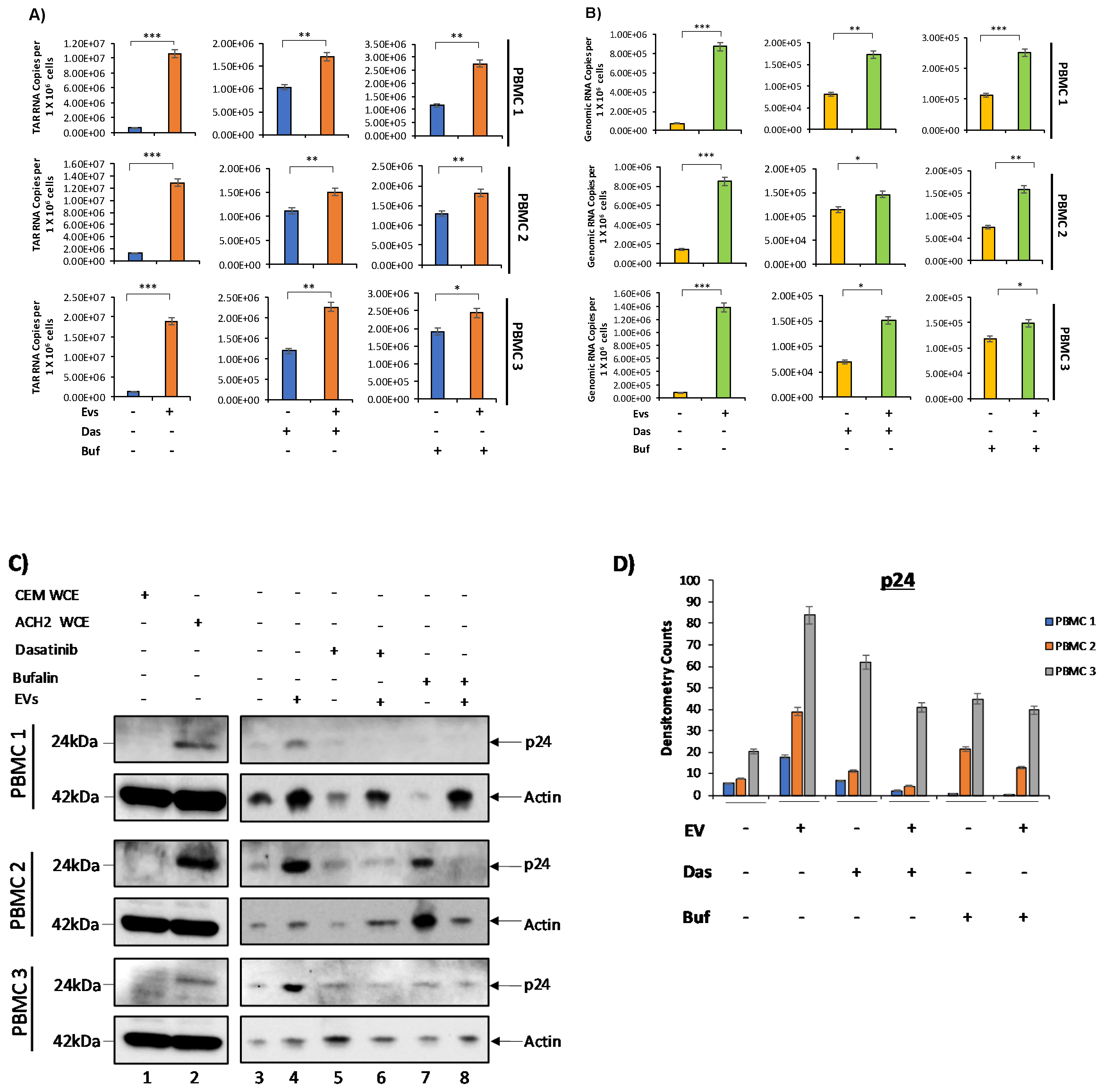
3.5. c-Src and SRC-1 Critical in HIV-1 Activation in Primary Cells
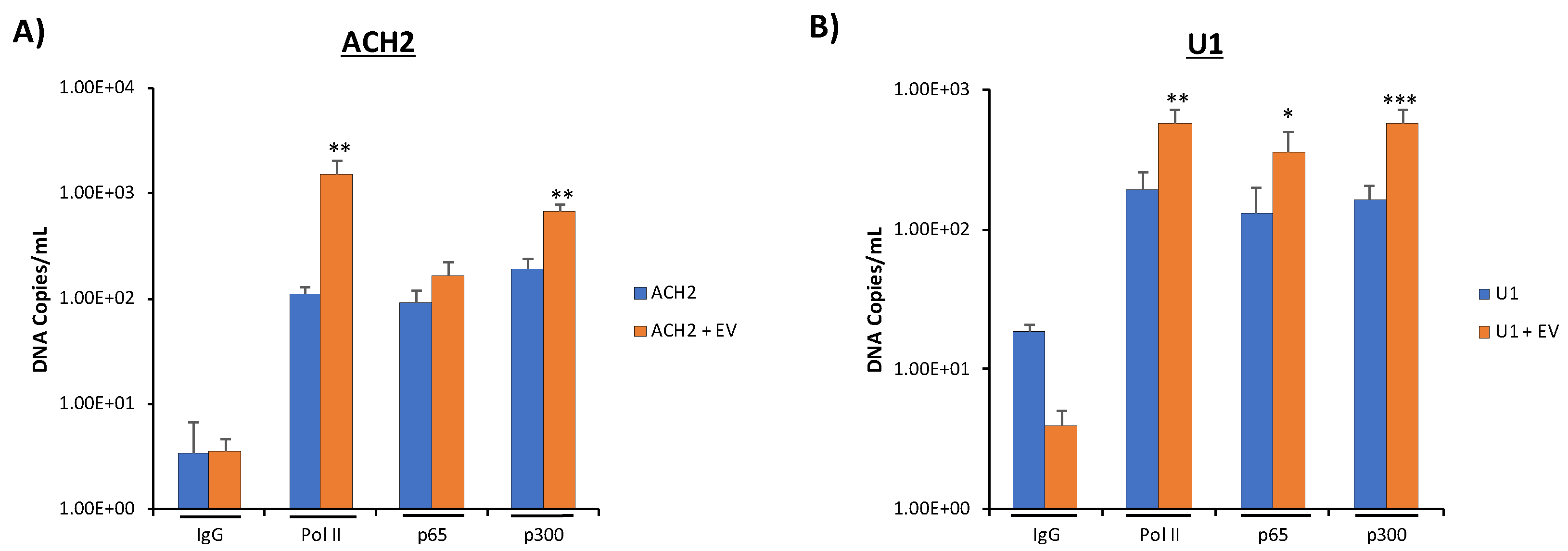
3.6. Increased Basal Transcription Is Driven by NF-κB/p300 Pathway
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ortblad, K.F.; Lozano, R.; Murray, C.J.L. The burden of HIV: Insights from the Global Burden of Disease Study 2010. AIDS 2013, 27, 2003–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Abbas, W.; Herbein, G. HIV-1 latency in monocytes/macrophages. Viruses 2014, 6, 1837–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, J.M. Latent HIV dynamics and implications for sustained viral suppression in the absence of antiretroviral therapy. J. Virus Erad. 2018, 4, 91–98. [Google Scholar]
- Barton, K.; Burch, B.; Soriano-Sarabia, N.; Margolis, D. Prospects for treatment of latent HIV. Clin. Pharmacol. Ther. 2013, 93, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisele, E.; Siliciano, R.F. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity 2012, 37, 377–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, G.R.; Pasquier, E.; Watkins, J.; Bourgarel-Rey, V.; Peyrot, V.; Esquieu, D.; Barbier, P.; de Mareuil, J.; Braguer, D.; Kaleebu, P.; et al. The Glutamine-rich Region of the HIV-1 Tat Protein Is Involved in T-cell Apoptosis. J. Biol. Chem. 2004, 279, 48197–48204. [Google Scholar] [CrossRef] [Green Version]
- Barclay, R.A.; Schwab, A.; DeMarino, C.; Akpamagbo, Y.; Lepene, B.; Kassaye, S.; Iordanskiy, S.; Kashanchi, F. Exosomes from uninfected cells activate transcription of latent HIV-1. J. Biol. Chem. 2017, 292, 11682–11701. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, R.M.; Mathew, A.; Mason, A.B.; Teng, K. Exosome formation during maturation of mammalian and avian reticulocytes: Evidence that exosome release is a major route for externalization of obsolete membrane proteins. J. Cell. Physiol. 1991, 147, 27–36. [Google Scholar] [CrossRef]
- Gu, Y.; Li, M.; Wang, T.; Liang, Y.; Zhong, Z.; Wang, X.; Zhou, Q.; Chen, L.; Lang, Q.; He, Z.; et al. Lactation-Related MicroRNA Expression Profiles of Porcine Breast Milk Exosomes. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Rashed, M.H.; Bayraktar, E.; Helal, G.K.; Abd-Ellah, M.F.; Amero, P.; Chavez-Reyes, A.; Rodriguez-Aguayo, C. Exosomes: From Garbage Bins to Promising Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 538. [Google Scholar] [CrossRef] [Green Version]
- Admyre, C.; Johansson, S.M.; Qazi, K.R.; Filén, J.-J.; Lahesmaa, R.; Norman, M.; Neve, E.P.A.; Scheynius, A.; Gabrielsson, S. Exosomes with Immune Modulatory Features Are Present in Human Breast Milk. J. Immunol. 2007, 179, 1969–1978. [Google Scholar] [CrossRef]
- Batagov, A.O.; Kurochkin, I.V. Exosomes secreted by human cells transport largely mRNA fragments that are enriched in the 3′-untranslated regions. Biol. Direct 2013, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleet, M.L.; DeMarino, C.; Lepene, B.; Aman, M.J.; Kashanchi, F. The Role of Exosomal VP40 in Ebola Virus Disease. DNA Cell Biol. 2017, 36, 243–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampey, G.C.; Meyering, S.S.; Zadeh, M.A.; Saifuddin, M.; Hakami, R.M.; Kashanchi, F. Exosomes and their role in CNS viral infections. J. Neurovirol. 2014, 20, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Ahsan, N.A.; Sampey, G.C.; Lepene, B.; Akpamagbo, Y.; Barclay, R.A.; Iordanskiy, S.; Hakami, R.M.; Kashanchi, F. Presence of Viral RNA and Proteins in Exosomes from Cellular Clones Resistant to Rift Valley Fever Virus Infection. Front. Microbiol. 2016, 7, 139. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.; Sampey, G.; Chung, M.-C.; Bailey, C.; Van Hoek, M.L.; Kashanchi, F.; Hakami, R.M. The carrying pigeons of the cell: Exosomes and their role in infectious diseases caused by human pathogens. Pathog. Dis. 2014, 71, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Alenquer, M.; Amorim, M.J. Exosome Biogenesis, Regulation, and Function in Viral Infection. Viruses 2015, 7, 5066–5083. [Google Scholar] [CrossRef]
- Kalani, A.; Tyagi, A.; Tyagi, N. Exosomes: Mediators of Neurodegeneration, Neuroprotection and Therapeutics. Mol. Neurobiol. 2014, 49, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, A.; Iordanskiy, S.; Das, R.; Van Duyne, R.; Santos, S.; Jaworski, E.; Guendel, I.; Sampey, G.; Dalby, E.; Iglesias-Ussel, M.; et al. Exosomes Derived from HIV-1-infected Cells Contain Trans-activation Response Element RNA. J. Biol. Chem. 2013, 288, 20014–20033. [Google Scholar] [CrossRef] [Green Version]
- DeMarino, C.; Pleet, M.L.; Cowen, M.; Barclay, R.A.; Akpamagbo, Y.; Erickson, J.; Ndembe, N.; Charurat, M.; Jumare, J.; Bwala, S.; et al. Antiretroviral Drugs Alter the Content of Extracellular Vesicles from HIV-1-Infected Cells. Sci. Rep. 2018, 8, 7653. [Google Scholar] [CrossRef] [PubMed]
- Akpamagbo, Y.A.; DeMarino, C.; Pleet, M.L.; Schwab, A.; Rodriguez, M.; Barclay, R.A.; Sampey, G.; Iordanskiy, S.; El-Hage, N.; Kashanchi, F. HIV-1 Transcription Inhibitors Increase the Synthesis of Viral Non-Coding RNA that Contribute to Latency. Curr. Pharm. Des. 2017, 23, 4133–4144. [Google Scholar] [CrossRef]
- Ung, T.H.; Madsen, H.J.; Hellwinkel, J.E.; Lencioni, A.M.; Graner, M.W. Exosome proteomics reveals transcriptional regulator proteins with potential to mediate downstream pathways. Cancer Sci. 2014, 105, 1384–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.M.; Brugge, J.S. Cellular Functions Regulated by Src Family Kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irby, R.B.; Yeatman, T.J. Role of Src expression and activation in human cancer. Oncogene 2000, 19, 5636–5642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maa, M.C.; Leu, T.H.; McCarley, D.J.; Schatzman, R.C.; Parsons, S.J. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: Implications for the etiology of multiple human cancers. Proc. Natl. Acad. Sci. USA 1995, 92, 6981–6985. [Google Scholar] [CrossRef] [Green Version]
- Biscardi, J.S.; Ishizawar, R.C.; Silva, C.M.; Parsons, S.J. Tyrosine kinase signalling in breast cancer: Epidermal growth factor receptor and c-Src interactions in breast cancer. Breast Cancer Res. 2000, 2, 203–210. [Google Scholar] [CrossRef] [Green Version]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [Green Version]
- LoPiccolo, J.; Blumenthal, G.M.; Bernstein, W.B.; Dennis, P.A. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist. Updates 2008, 11, 32–50. [Google Scholar] [CrossRef] [Green Version]
- Heredia, A.; Le, N.; Gartenhaus, R.B.; Sausville, E.; Medina-Moreno, S.; Zapata, J.C.; Davis, C.; Gallo, R.C.; Redfield, R.R. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9412–9417. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, S.D.S.; Sakac, D.; Neschadim, A.; Branch, D.R. c-SRC protein tyrosine kinase regulates early HIV-1 infection post-entry. AIDS 2016, 30, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Gallay, P.; Swingler, S.; Aiken, C.; Trono, D. HIV-1 infection of nondividing cells: C-terminal tyrosine phosphorylation of the viral matrix protein is a key regulator. Cell 1995, 80, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Harmon, B.; Campbell, N.; Ratner, L. Role of Abl Kinase and the Wave2 Signaling Complex in HIV-1 Entry at a Post-Hemifusion Step. PLoS Pathog. 2010, 6, e1000956. [Google Scholar] [CrossRef] [Green Version]
- Readinger, J.A.; Schiralli, G.M.; Jiang, J.-K.; Thomas, C.J.; August, A.; Henderson, A.J.; Schwartzberg, P.L. Selective targeting of ITK blocks multiple steps of HIV replication. Proc. Natl. Acad. Sci. USA 2008, 105, 6684–6689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Molecular Med. 2005, 9, 59–71. [Google Scholar] [CrossRef]
- Ersahin, T.; Tuncbag, N.; Cetin-Atalay, R. The PI3K/AKT/mTOR interactive pathway. Mol. BioSyst. 2015, 11, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Yao, S.; Hu, M.; Li, W.; Hao, T.; Zhou, F.; Zhu, X.; Lu, H.; Qin, D.; Yan, Q.; et al. HIV-1 Nef and KSHV oncogene K1 synergistically promote angiogenesis by inducing cellular miR-718 to regulate the PTEN/AKT/mTOR signaling pathway. Nucleic Acids Res. 2014, 42, 9862–9879. [Google Scholar] [CrossRef] [PubMed]
- Sampey, G.C.; Saifuddin, M.; Schwab, A.; Barclay, R.; Punya, S.; Chung, M.-C.; Hakami, R.M.; Zadeh, M.A.; Lepene, B.; Klase, Z.A.; et al. Exosomes from HIV-1-infected Cells Stimulate Production of Pro-inflammatory Cytokines through Trans-activating Response (TAR) RNA. J. Biol. Chem. 2016, 291, 1251–1266. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, C.; Barat, C.; Cantin, R.; Tremblay, M.J. Involvement of Src and Syk Tyrosine Kinases in HIV-1 Transfer from Dendritic Cells to CD4+ T Lymphocytes. J. Immunol. 2007, 178, 2862–2871. [Google Scholar] [CrossRef] [Green Version]
- Trible, R.P.; Emert-Sedlak, L.; Smithgall, T.E. HIV-1 Nef Selectively Activates Src Family Kinases Hck, Lyn, and c-Src through Direct SH3 Domain Interaction. J. Biol. Chem. 2006, 281, 27029–27038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Why the Need and How to Approach the Functional Diversity of Extracellular Vesicles. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5717434/ (accessed on 3 January 2020).
- Walker, V.G.; Ammer, A.; Cao, Z.; Clump, A.C.; Jiang, B.-H.; Kelley, L.C.; Weed, S.A.; Zot, H.; Flynn, D.C. PI3K activation is required for PMA-directed activation of cSrc by AFAP-110. Am. J. Physiol. Cell Physiol. 2007, 293, C119–C132. [Google Scholar] [CrossRef]
- Amata, I.; Maffei, M.; Pons, M. Phosphorylation of unique domains of Src family kinases. Front. Genet 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.Z.; Thuraisingam, T.; Kanagaratham, C.; Tao, S.; Radzioch, D. c-Src kinase is involved in the tyrosine phosphorylation and activity of SLC11A1 in differentiating macrophages. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Glazer, R.I. Purification and characterization of p93fes- and p60src-related tyrosine protein kinase activities in differentiated HL-60 leukemia cells. J. Biol. Chem. 1987, 262, 17543–17548. [Google Scholar]
- Sato, K.; Sato, A.; Aoto, M.; Fukami, Y. c-Src phosphorylates epidermal growth factor receptor on tyrosine 845. Biochem. Biophys. Res. Commun. 1995, 215, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. [Google Scholar] [CrossRef] [Green Version]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Freudlsperger, C.; Burnett, J.R.; Friedman, J.A.; Kannabiran, V.R.; Chen, Z.; Van Waes, C. EGFR-PI3K-AKT-mTOR signaling in head and neck squamous cell carcinomas: Attractive targets for molecular-oriented therapy. Expert Opin. Ther. Targets 2011, 15, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E.; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.-L.; Guan, Y.-J.; Wang, L.; Wei, W.; Kane, A.B.; Chin, Y.E. Central role of the threonine residue within the p+1 loop of receptor tyrosine kinase in STAT3 constitutive phosphorylation in metastatic cancer cells. Mol. Cell. Biol. 2004, 24, 9390–9400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephanou, A.; Latchman, D.S. Opposing actions of STAT-1 and STAT-3. Growth Factors 2005, 23, 177–182. [Google Scholar] [CrossRef]
- Giraud, S.; Bienvenu, F.; Avril, S.; Gascan, H.; Heery, D.M.; Coqueret, O. Functional interaction of STAT3 transcription factor with the coactivator NcoA/SRC1a. J. Biol. Chem. 2002, 277, 8004–8011. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.A.; Qin, L.; Tien, J.C.-Y.; Young, L.S.; Xu, J. The function of steroid receptor coactivator-1 in normal tissues and cancer. Int. J. Biol. Sci. 2012, 8, 470–485. [Google Scholar] [CrossRef] [PubMed]
- Lindauer, M.; Hochhaus, A. Dasatinib. Recent Results Cancer Res. 2014, 201, 27–65. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, C.W. The Akt/PKB family of protein kinases: A review of small molecule inhibitors and progress towards target validation: A 2009 update. Curr. Top. Med. Chem. 2010, 10, 458–477. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Lonard, D.M.; Yu, Y.; Chow, D.-C.; Palzkill, T.G.; Wang, J.; Qi, R.; Matzuk, A.J.; Song, X.; Madoux, F.; et al. Bufalin is a potent small-molecule inhibitor of the steroid receptor coactivators SRC-3 and SRC-1. Cancer Res. 2014, 74, 1506–1517. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Chen, N.; Zhou, X.; Ge, X.; Feng, L.; Li, P.; Li, X.; Geng, L.; Wang, X. The STAT3 inhibitor WP1066 synergizes with vorinostat to induce apoptosis of mantle cell lymphoma cells. Biochem. Biophys. Res. Commun. 2015, 464, 292–298. [Google Scholar] [CrossRef]
- Nika, K.; Soldani, C.; Salek, M.; Paster, W.; Gray, A.; Etzensperger, R.; Fugger, L.; Polzella, P.; Cerundolo, V.; Dushek, O.; et al. Constitutively Active Lck Kinase in T Cells Drives Antigen Receptor Signal Transduction. Immunity 2010, 32, 766–777. [Google Scholar] [CrossRef] [Green Version]
- Masiello, D.; Gorospe, G.; Yang, A.S. The occurrence and management of fluid retention associated with TKI therapy in CML, with a focus on dasatinib. J. Hematol. Oncol. 2009, 2, 46. [Google Scholar] [CrossRef] [Green Version]
- Archin, N.M.; Sung, J.M.; Garrido, C.; Soriano-Sarabia, N.; Margolis, D.M. Eradicating HIV-1 infection: Seeking to clear a persistent pathogen. Nat. Rev. Microbiol. 2014, 12, 750–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lusic, M.; Marcello, A.; Cereseto, A.; Giacca, M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003, 22, 6550–6561. [Google Scholar] [CrossRef] [Green Version]
- Vo, N.; Goodman, R.H. CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem. 2001, 276, 13505–13508. [Google Scholar] [CrossRef] [Green Version]
- Mantelingu, K.; Reddy, B.A.A.; Swaminathan, V.; Kishore, A.H.; Siddappa, N.B.; Kumar, G.V.P.; Nagashankar, G.; Natesh, N.; Roy, S.; Sadhale, P.P.; et al. Specific inhibition of p300-HAT alters global gene expression and represses HIV replication. Chem. Biol. 2007, 14, 645–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetel, M.J. Nuclear receptor coactivators: Essential players for steroid hormone action in the brain and in behaviour. J. Neuroendocrinol. 2009, 21, 229–237. [Google Scholar] [CrossRef]
- Schwab, A.; Meyering, S.S.; Lepene, B.; Iordanskiy, S.; Van Hoek, M.L.; Hakami, R.M.; Kashanchi, F. Extracellular vesicles from infected cells: Potential for direct pathogenesis. Front. Microbiol. 2015, 6, 1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlassov, A.V.; Magdaleno, S.; Setterquist, R.; Conrad, R. Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochimica et Biophysica Acta (BBA)—General Subjects 2012, 1820, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Prieto, D.; Sotelo, N.; Seija, N.; Sernbo, S.; Abreu, C.; Durán, R.; Gil, M.; Sicco, E.; Irigoin, V.; Oliver, C.; et al. S100-A9 protein in exosomes from chronic lymphocytic leukemia cells promotes NF-κB activity during disease progression. Blood 2017, 130, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Konadu, K.A.; Chu, J.; Huang, M.B.; Amancha, P.K.; Armstrong, W.; Powell, M.D.; Villinger, F.; Bond, V.C. Association of Cytokines With Exosomes in the Plasma of HIV-1-Seropositive Individuals. J. Infect. Dis. 2015, 211, 1712–1716. [Google Scholar] [CrossRef]
- Litterst, C.M.; Pfitzner, E. Transcriptional activation by STAT6 requires the direct interaction with NCoA-1. J. Biol. Chem. 2001, 276, 45713–45721. [Google Scholar] [CrossRef] [Green Version]
- Marzio, G.; Tyagi, M.; Gutierrez, M.I.; Giacca, M. HIV-1 tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc. Natl. Acad. Sci. USA 1998, 95, 13519–13524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benkirane, M.; Chun, R.F.; Xiao, H.; Ogryzko, V.V.; Howard, B.H.; Nakatani, Y.; Jeang, K.T. Activation of integrated provirus requires histone acetyltransferase. p300 and P/CAF are coactivators for HIV-1 Tat. J. Biol. Chem. 1998, 273, 24898–24905. [Google Scholar] [CrossRef] [Green Version]
- Hatano, H.; Jain, V.; Hunt, P.W.; Lee, T.-H.; Sinclair, E.; Do, T.D.; Hoh, R.; Martin, J.N.; McCune, J.M.; Hecht, F.; et al. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. J. Infect. Dis. 2013, 208, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, H.; Dewar, R.L.; Adelsberger, J.W.; Rehm, C.A.; O’Doherty, U.; Paxinos, E.E.; Fauci, A.S.; Lane, H.C. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2016, 113, 8783–8788. [Google Scholar] [CrossRef] [Green Version]
- Maldarelli, F.; Kearney, M.; Palmer, S.; Stephens, R.; Mican, J.; Polis, M.A.; Davey, R.T.; Kovacs, J.; Shao, W.; Rock-Kress, D.; et al. HIV populations are large and accumulate high genetic diversity in a nonlinear fashion. J. Virol. 2013, 87, 10313–10323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boltz, V.F.; Zheng, Y.; Lockman, S.; Hong, F.; Halvas, E.K.; McIntyre, J.; Currier, J.S.; Chibowa, M.C.; Kanyama, C.; Nair, A.; et al. Role of low-frequency HIV-1 variants in failure of nevirapine-containing antiviral therapy in women previously exposed to single-dose nevirapine. Proc. Natl. Acad. Sci. USA 2011, 108, 9202–9207. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.M.; Borodowsky, I.; Fernandez, B.; Gonzalez, L.; Kumar, M. Human immunodeficiency virus type 1 RNA Levels in different regions of human brain: Quantification using real-time reverse transcriptase-polymerase chain reaction. J. Neurovirol. 2007, 13, 210–224. [Google Scholar] [CrossRef]
- Cihlar, T.; Fordyce, M. Current status and prospects of HIV treatment. Curr. Opin. Virol. 2016, 18, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef]
- Honda, S.; Sadatomi, D.; Yamamura, Y.; Nakashioya, K.; Tanimura, S.; Takeda, K. WP1066 suppresses macrophage cell death induced by inflammasome agonists independently of its inhibitory effect on STAT3. Cancer Sci. 2017, 108, 520–527. [Google Scholar] [CrossRef] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barclay, R.A.; Mensah, G.A.; Cowen, M.; DeMarino, C.; Kim, Y.; Pinto, D.O.; Erickson, J.; Kashanchi, F. Extracellular Vesicle Activation of Latent HIV-1 Is Driven by EV-Associated c-Src and Cellular SRC-1 via the PI3K/AKT/mTOR Pathway. Viruses 2020, 12, 665. https://doi.org/10.3390/v12060665
Barclay RA, Mensah GA, Cowen M, DeMarino C, Kim Y, Pinto DO, Erickson J, Kashanchi F. Extracellular Vesicle Activation of Latent HIV-1 Is Driven by EV-Associated c-Src and Cellular SRC-1 via the PI3K/AKT/mTOR Pathway. Viruses. 2020; 12(6):665. https://doi.org/10.3390/v12060665
Chicago/Turabian StyleBarclay, Robert A., Gifty A. Mensah, Maria Cowen, Catherine DeMarino, Yuriy Kim, Daniel O. Pinto, James Erickson, and Fatah Kashanchi. 2020. "Extracellular Vesicle Activation of Latent HIV-1 Is Driven by EV-Associated c-Src and Cellular SRC-1 via the PI3K/AKT/mTOR Pathway" Viruses 12, no. 6: 665. https://doi.org/10.3390/v12060665
APA StyleBarclay, R. A., Mensah, G. A., Cowen, M., DeMarino, C., Kim, Y., Pinto, D. O., Erickson, J., & Kashanchi, F. (2020). Extracellular Vesicle Activation of Latent HIV-1 Is Driven by EV-Associated c-Src and Cellular SRC-1 via the PI3K/AKT/mTOR Pathway. Viruses, 12(6), 665. https://doi.org/10.3390/v12060665