Extracellular Vesicles in Viral Spread and Antiviral Response


{kind=link}
Abstract
:1. Introduction
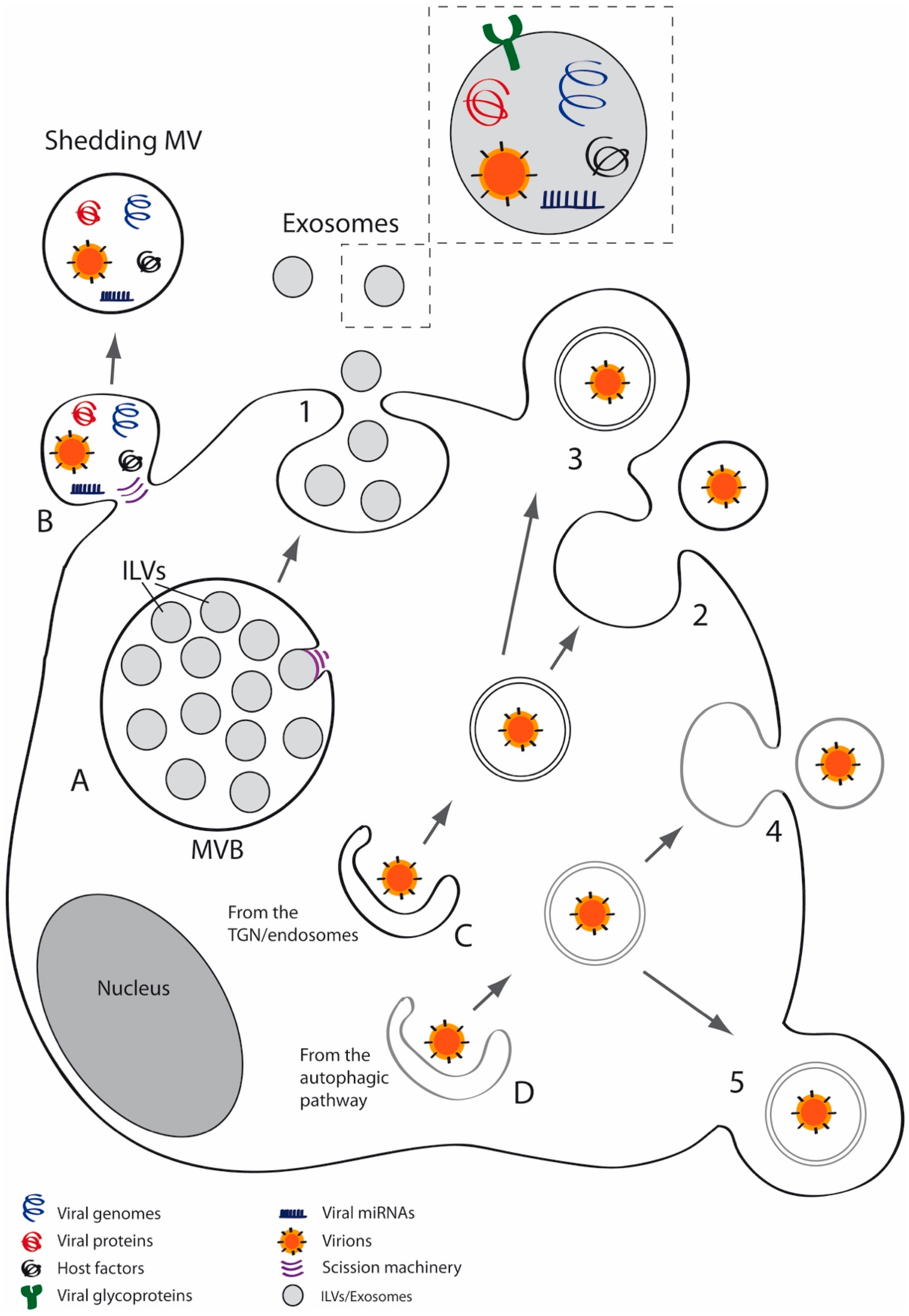
2. EVs: Brief Overview
3. EVs in Viral Infections
3.1. Non-Enveloped Viruses
3.2. Enveloped Viruses
4. EVs in Herpesvirus Infections
4.1. Alphaherpesviruses
4.2. Betaherpesviruses
4.3. Gammaherpesviruses
5. The Role of Lipid Rafts and the MAL Proteolipid
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sanjuan, R.; Thoulouze, M.I. Why viruses sometimes disperse in groups. Virus Evol. 2019, 5, vez014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.R.; Kashanchi, F.; Jacobson, S. Exosomes in Viral Disease. Neurotherapeutics 2016, 13, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.W.; Lichty, B.D.; Bowdish, D.M. Microvesicles: Ubiquitous contributors to infection and immunity. J. Leukoc. Biol. 2015, 97, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr. Exosomal communication goes viral. J. Virol. 2015, 89, 5200–5203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alenquer, M.; Amorim, M.J. Exosome Biogenesis, Regulation, and Function in Viral Infection. Viruses 2015, 7, 5066–5083. [Google Scholar] [CrossRef] [PubMed]
- Wurdinger, T.; Gatson, N.N.; Balaj, L.; Kaur, B.; Breakefield, X.O.; Pegtel, D.M. Extracellular vesicles and their convergence with viral pathways. Adv. Virol. 2012, 2012, 767694. [Google Scholar] [CrossRef]
- Schwab, A.; Meyering, S.S.; Lepene, B.; Iordanskiy, S.; van Hoek, M.L.; Hakami, R.M.; Kashanchi, F. Extracellular vesicles from infected cells: Potential for direct pathogenesis. Front. Microbiol. 2015, 6, 1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schorey, J.S.; Cheng, Y.; Singh, P.P.; Smith, V.L. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep. 2015, 16, 24–43. [Google Scholar] [CrossRef] [Green Version]
- Meckes, D.G., Jr.; Raab-Traub, N. Microvesicles and viral infection. J. Virol. 2011, 85, 12844–12854. [Google Scholar] [CrossRef] [Green Version]
- Van Dongen, H.M.; Masoumi, N.; Witwer, K.W.; Pegtel, D.M. Extracellular Vesicles Exploit Viral Entry Routes for Cargo Delivery. Microbiol. Mol. Biol. Rev. Membr. 2016, 80, 369–386. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, V.L.; Ellwanger, J.H.; Chies, J.A.B. Extracellular vesicles in host-pathogen interactions and immune regulation—Exosomes as emerging actors in the immunological theater of pregnancy. Heliyon 2019, 5, e02355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreu-Moreno, I.; Sanjuan, R. Collective Viral Spread Mediated by Virion Aggregates Promotes the Evolution of Defective Interfering Particles. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altan-Bonnet, N.; Perales, C.; Domingo, E. Extracellular vesicles: Vehicles of en bloc viral transmission. Virus Res. 2019, 265, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Leeks, A.; Sanjuan, R.; West, S.A. The evolution of collective infectious units in viruses. Virus Res. 2019, 265, 94–101. [Google Scholar] [CrossRef]
- Altan-Bonnet, N. Extracellular vesicles are the Trojan horses of viral infection. Curr. Opin. Microbiol. 2016, 32, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Kouwaki, T.; Okamoto, M.; Tsukamoto, H.; Fukushima, Y.; Oshiumi, H. Extracellular Vesicles Deliver Host and Virus RNA and Regulate Innate Immune Response. Int. J. Mol. Sci. 2017, 18, 666. [Google Scholar] [CrossRef] [Green Version]
- Urbanelli, L.; Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Porcellati, S.; Emiliani, C. The Role of Extracellular Vesicles in Viral Infection and Transmission. Vaccines 2019, 7, 102. [Google Scholar] [CrossRef] [Green Version]
- Crenshaw, B.J.; Gu, L.; Sims, B.; Matthews, Q.L. Exosome Biogenesis and Biological Function in Response to Viral Infections. Open Virol. J. 2018, 12, 134–148. [Google Scholar] [CrossRef] [Green Version]
- Raab-Traub, N.; Dittmer, D.P. Viral effects on the content and function of extracellular vesicles. Nat. Rev. Microbiol. 2017, 15, 559–572. [Google Scholar] [CrossRef]
- Nolte-’t Hoen, E.; Cremer, T.; Gallo, R.C.; Margolis, L.B. Extracellular vesicles and viruses: Are they close relatives? Proc. Natl. Acad. Sci. USA 2016, 113, 9155–9161. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.C.; Huber, M.T. Directed egress of animal viruses promotes cell-to-cell spread. J. Virol. 2002, 76, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, M.; Generous, A.; Sinn, P.L.; Cattaneo, R. Connections matter—How viruses use cell-cell adhesion components. J. Cell Sci. 2015, 128, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graw, F.; Perelson, A.S. Modeling Viral Spread. Annu. Rev. Virol. 2016, 3, 555–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sin, J.; Mangale, V.; Thienphrapa, W.; Gottlieb, R.A.; Feuer, R. Recent progress in understanding coxsackievirus replication, dissemination, and pathogenesis. Virology 2015, 484, 288–304. [Google Scholar] [CrossRef] [Green Version]
- Weed, D.J.; Nicola, A.V. Herpes simplex virus Membrane Fusion. Adv. Anat. Embryol. Cell Biol. 2017, 223, 29–47. [Google Scholar]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuana, Y.; Sturk, A.; Nieuwland, R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013, 27, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Sedgwick, A.E.; D’Souza-Schorey, C. The biology of extracellular microvesicles. Traffic 2018, 19, 319–327. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.L.N.; Breakefield, X.O.; Weaver, A.M. Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends Cell Biol. 2017, 27, 172–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budnik, V.; Ruiz-Canada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 2016, 17, 160–172. [Google Scholar] [CrossRef] [Green Version]
- Del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; Lopez, J.A. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef]
- Wei, X.; Liu, C.; Wang, H.; Wang, L.; Xiao, F.; Guo, Z.; Zhang, H. Surface Phosphatidylserine Is Responsible for the Internalization on Microvesicles Derived from Hypoxia-Induced Human Bone Marrow Mesenchymal Stem Cells into Human Endothelial Cells. PLoS ONE 2016, 11, e0147360. [Google Scholar] [CrossRef] [Green Version]
- Scott, S.; Pendlebury, S.A.; Green, C. Lipid organization in erythrocyte membrane microvesicles. Biochem. J. 1984, 224, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Bello-Morales, R.; Lopez-Guerrero, J.A. Isolation/Analysis of Extracellular Microvesicles from HSV-1-Infected Cells. In Herpes Simplex Virus; Springer Nature: Basel, Switzerland, 2020; Volume 2060, pp. 305–317. [Google Scholar]
- Witwer, K.W.; Buzas, E.I.; Bemis, L.T.; Bora, A.; Lasser, C.; Lotvall, J.; Nolte-’t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2, 20360. [Google Scholar] [CrossRef]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Gould, S.J.; Raposo, G. As we wait: Coping with an imperfect nomenclature for extracellular vesicles. J. Extracell. Vesicles 2013, 2, 20389. [Google Scholar] [CrossRef]
- Lane, R.E.; Korbie, D.; Trau, M.; Hill, M.M. Purification Protocols for Extracellular Vesicles. Methods Mol. Biol. 2017, 1660, 111–130. [Google Scholar] [PubMed] [Green Version]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Pol, E.; Boing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharm. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyorgy, B.; Szabo, T.G.; Pasztoi, M.; Pal, Z.; Misjak, P.; Aradi, B.; Laszlo, V.; Pallinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros, F.M.; Carneiro, F.; Machado, J.C.; Melo, S.A. Exosomes and Immune Response in Cancer: Friends or Foes? Front. Immunol. 2018, 9, 730. [Google Scholar] [CrossRef] [PubMed]
- Nogues, L.; Benito-Martin, A.; Hergueta-Redondo, M.; Peinado, H. The influence of tumour-derived extracellular vesicles on local and distal metastatic dissemination. Mol. Asp. Med. 2018, 60, 15–26. [Google Scholar] [CrossRef]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer—Implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Clancy, J.W.; Sedgwick, A.; D’Souza-Schorey, C. Microvesicles: Mediators of extracellular communication during cancer progression. J. Cell Sci. 2010, 123 Pt 10, 1603–1611. [Google Scholar] [CrossRef] [Green Version]
- Silverman, J.M.; Reiner, N.E. Exosomes and other microvesicles in infection biology: Organelles with unanticipated phenotypes. Cell. Microbiol. 2011, 13, 1–9. [Google Scholar] [CrossRef]
- Robbins, P.D.; Dorronsoro, A.; Booker, C.N. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J. Clin. Investig. 2016, 126, 1173–1180. [Google Scholar] [CrossRef] [Green Version]
- Fruhbeis, C.; Frohlich, D.; Kuo, W.P.; Amphornrat, J.; Thilemann, S.; Saab, A.S.; Kirchhoff, F.; Mobius, W.; Goebbels, S.; Nave, K.A.; et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 2013, 11, e1001604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pusic, K.M.; Pusic, A.D.; Kraig, R.P. Environmental Enrichment Stimulates Immune Cell Secretion of Exosomes that Promote CNS Myelination and May Regulate Inflammation. Cell Mol. Neurobiol. 2016, 36, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, M.M.; Kaiser, J.; Schwab, M.E. Extracellular Vesicles: Multimodal Envoys in Neural Maintenance and Repair. Trends Neurosci. 2018, 41, 360–372. [Google Scholar] [CrossRef]
- Lopez-Leal, R.; Court, F.A. Schwann Cell Exosomes Mediate Neuron-Glia Communication and Enhance Axonal Regeneration. Cell. Mol. Neurobiol. 2016, 36, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Basso, M.; Bonetto, V. Extracellular Vesicles and a Novel Form of Communication in the Brain. Front. Neurosci. 2016, 10, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Ma, P.; Deng, L.; Liu, Z.; Wang, X.; Liu, X.; Long, G. Hepatitis A virus structural protein pX interacts with ALIX and promotes the secretion of virions and foreign proteins through exosome-like vesicles. J. Extracell. Vesicles 2020, 9, 1716513. [Google Scholar] [CrossRef]
- Nagashima, S.; Jirintai, S.; Takahashi, M.; Kobayashi, T.; Tanggis; Nishizawa, T.; Kouki, T.; Yashiro, T.; Okamoto, H. Hepatitis E virus egress depends on the exosomal pathway, with secretory exosomes derived from multivesicular bodies. J. Gen. Virol. 2014, 95 Pt 10, 2166–2175. [Google Scholar] [CrossRef]
- Feng, Z.; Hirai-Yuki, A.; McKnight, K.L.; Lemon, S.M. Naked Viruses That Aren’t Always Naked: Quasi-Enveloped Agents of Acute Hepatitis. Annu. Rev. Virol. 2014, 1, 539–560. [Google Scholar] [CrossRef]
- Taylor, M.P.; Burgon, T.B.; Kirkegaard, K.; Jackson, W.T. Role of microtubules in extracellular release of poliovirus. J. Virol. 2009, 83, 6599–6609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackwell, J.H.; Wool, S.; Kosikowski, F.V. Vesicular exocytosis of foot- and -mouth disease virus from mammary gland secretory epithelium of infected cows. J. Gen. Virol. 1981, 56 Pt 1, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Xu, S.; Shi, X.; Xu, G.; Shen, C.; Liu, X.; Zheng, H. Exosomes-mediated transmission of foot-and-mouth disease virus in vivo and in vitro. Vet. Microbiol. 2019, 233, 164–173. [Google Scholar] [CrossRef]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Hancock, B.M.; et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef] [PubMed]
- Sin, J.; McIntyre, L.; Stotland, A.; Feuer, R.; Gottlieb, R.A. Coxsackievirus B Escapes the Infected Cell in Ejected Mitophagosomes. J. Virol. 2017, 91, e01347-17. [Google Scholar] [CrossRef] [Green Version]
- Germano, J.F.; Sawaged, S.; Saadaeijahromi, H.; Andres, A.M.; Feuer, R.; Gottlieb, R.A.; Sin, J. Coxsackievirus B infection induces the extracellular release of miR-590-5p, a proviral microRNA. Virology 2019, 529, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Wu, J.; Shen, L.; Yang, J.; Chen, J.; Xu, H. Enterovirus 71 transmission by exosomes establishes a productive infection in human neuroblastoma cells. Virus Genes 2016, 52, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Wu, J.; Fang, D.; Qiu, Y.; Zou, X.; Jia, X.; Yin, Y.; Shen, L.; Mao, L. Exosomes cloak the virion to transmit Enterovirus 71 non-lytically. Virulence 2020, 11, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Van der Grein, S.G.; Defourny, K.A.Y.; Rabouw, H.H.; Galiveti, C.R.; Langereis, M.A.; Wauben, M.H.M.; Arkesteijn, G.J.A.; van Kuppeveld, F.J.M.; Nolte-’t Hoen, E.N.M. Picornavirus infection induces temporal release of multiple extracellular vesicle subsets that differ in molecular composition and infectious potential. Plos Pathog. 2019, 15, e1007594. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, B.A.; Morris-Love, J.; Gee, G.V.; Haley, S.A.; Atwood, W.J. JC Virus infected choroid plexus epithelial cells produce extracellular vesicles that infect glial cells independently of the virus attachment receptor. PLoS Pathog. 2020, 16, e1008371. [Google Scholar] [CrossRef]
- Ranjit, S.; Kodidela, S.; Sinha, N.; Chauhan, S.; Kumar, S. Extracellular Vesicles from Human Papilloma Virus-Infected Cervical Cancer Cells Enhance HIV-1 Replication in Differentiated U1 Cell Line. Viruses 2020, 12, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiana, M.; Ghosh, S.; Ho, B.A.; Rajasekaran, V.; Du, W.L.; Mutsafi, Y.; De Jesus-Diaz, D.A.; Sosnovtsev, S.V.; Levenson, E.A.; Parra, G.I.; et al. Vesicle-Cloaked Virus Clusters Are Optimal Units for Inter-organismal Viral Transmission. Cell Host Microbe 2018, 24, 208–220 e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirblich, C.; Bhattacharya, B.; Roy, P. Nonstructural protein 3 of bluetongue virus assists virus release by recruiting ESCRT-I protein Tsg101. J. Virol. 2006, 80, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celma, C.C.; Roy, P. A viral nonstructural protein regulates bluetongue virus trafficking and release. J. Virol. 2009, 83, 6806–6816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, B.; Celma, C.C.; Roy, P. Influence of cellular trafficking pathway on bluetongue virus infection in ovine cells. Viruses 2015, 7, 2378–2403. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef] [Green Version]
- Bukong, T.N.; Momen-Heravi, F.; Kodys, K.; Bala, S.; Szabo, G. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog. 2014, 10, e1004424. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Zhou, J.; Wang, H. Host microRNAs and exosomes that modulate influenza virus infection. Virus Res. 2020, 279, 197885. [Google Scholar] [CrossRef]
- Maemura, T.; Fukuyama, S.; Kawaoka, Y. High Levels of miR-483-3p Are Present in Serum Exosomes Upon Infection of Mice With Highly Pathogenic Avian Influenza Virus. Front. Microbiol. 2020, 11, 144. [Google Scholar] [CrossRef]
- Liu, Y.M.; Tseng, C.H.; Chen, Y.C.; Yu, W.Y.; Ho, M.Y.; Ho, C.Y.; Lai, M.M.C.; Su, W.C. Exosome-delivered and Y RNA-derived small RNA suppresses influenza virus replication. J. Biomed. Sci. 2019, 26, 58. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Lata, S.; Ali, A.; Banerjea, A.C. Dengue haemorrhagic fever: A job done via exosomes? Emerg. Microbes Infect. 2019, 8, 1626–1635. [Google Scholar] [CrossRef] [Green Version]
- Freitas, M.N.; Marten, A.D.; Moore, G.A.; Tree, M.O.; McBrayer, S.P.; Conway, M.J. Extracellular vesicles restrict dengue virus fusion in Aedes aegypti cells. Virology 2020, 541, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Velandia-Romero, M.L.; Calderon-Pelaez, M.A.; Balbas-Tepedino, A.; Marquez-Ortiz, R.A.; Madronero, L.J.; Barreto Prieto, A.; Castellanos, J.E. Extracellular vesicles of U937 macrophage cell line infected with DENV-2 induce activation in endothelial cells EA.hy926. PLoS ONE 2020, 15, e0227030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Rojas, P.P.; Quiroz-Garcia, E.; Monroy-Martinez, V.; Agredano-Moreno, L.T.; Jimenez-Garcia, L.F.; Ruiz-Ordaz, B.H. Participation of Extracellular Vesicles from Zika-Virus-Infected Mosquito Cells in the Modification of Naive Cells’ Behavior by Mediating Cell-to-Cell Transmission of Viral Elements. Cells 2020, 9, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madison, M.N.; Okeoma, C.M. Exosomes: Implications in HIV-1 Pathogenesis. Viruses 2015, 7, 4093–4118. [Google Scholar] [CrossRef] [Green Version]
- Ellwanger, J.H.; Veit, T.D.; Chies, J.A.B. Exosomes in HIV infection: A review and critical look. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2017, 53, 146–154. [Google Scholar] [CrossRef]
- Felli, C.; Vincentini, O.; Silano, M.; Masotti, A. HIV-1 Nef Signaling in Intestinal Mucosa Epithelium Suggests the Existence of an Active Inter-kingdom Crosstalk Mediated by Exosomes. Front. Microbiol. 2017, 8, 1022. [Google Scholar] [CrossRef]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotic, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitas, A.; Peterlin, B.M. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef]
- Pereira, E.A.; daSilva, L.L. HIV-1 Nef: Taking Control of Protein Trafficking. Traffic 2016, 17, 976–996. [Google Scholar] [CrossRef] [Green Version]
- McNamara, R.P.; Costantini, L.M.; Myers, T.A.; Schouest, B.; Maness, N.J.; Griffith, J.D.; Damania, B.A.; MacLean, A.G.; Dittmer, D.P. Nef Secretion into Extracellular Vesicles or Exosomes Is Conserved across Human and Simian Immunodeficiency Viruses. mBio 2018, 9, e02344-17. [Google Scholar] [CrossRef] [Green Version]
- Arenaccio, C.; Anticoli, S.; Manfredi, F.; Chiozzini, C.; Olivetta, E.; Federico, M. Latent HIV-1 is activated by exosomes from cells infected with either replication-competent or defective HIV-1. Retrovirology 2015, 12, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, S.J.; Booth, A.M.; Hildreth, J.E. The Trojan exosome hypothesis. Proc. Natl. Acad. Sci. USA 2003, 100, 10592–10597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.G.; Booth, A.; Gould, S.J.; Hildreth, J.E. Evidence that HIV budding in primary macrophages occurs through the exosome release pathway. J. Biol. Chem. 2003, 278, 52347–52354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coren, L.V.; Shatzer, T.; Ott, D.E. CD45 immunoaffinity depletion of vesicles from Jurkat T cells demonstrates that exosomes contain CD45: No evidence for a distinct exosome/HIV-1 budding pathway. Retrovirology 2008, 5, 64. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo-Useros, N.; Naranjo-Gomez, M.; Archer, J.; Hatch, S.C.; Erkizia, I.; Blanco, J.; Borras, F.E.; Puertas, M.C.; Connor, J.H.; Fernandez-Figueras, M.T.; et al. Capture and transfer of HIV-1 particles by mature dendritic cells converges with the exosome-dissemination pathway. Blood 2009, 113, 2732–2741. [Google Scholar] [CrossRef] [Green Version]
- Hildreth, J.E.K. HIV As Trojan Exosome: Immunological Paradox Explained? Front. Immunol. 2017, 8, 1715. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo-Useros, N.; Naranjo-Gomez, M.; Erkizia, I.; Puertas, M.C.; Borras, F.E.; Blanco, J.; Martinez-Picado, J. HIV and mature dendritic cells: Trojan exosomes riding the Trojan horse? Plos Pathog. 2010, 6, e1000740. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.O.; DeMarino, C.; Pleet, M.L.; Cowen, M.; Branscome, H.; Al Sharif, S.; Jones, J.; Dutartre, H.; Lepene, B.; Liotta, L.A.; et al. HTLV-1 Extracellular Vesicles Promote Cell-to-Cell Contact. Front. Microbiol. 2019, 10, 2147. [Google Scholar] [CrossRef]
- Gunasekaran, M.; Bansal, S.; Ravichandran, R.; Sharma, M.; Perincheri, S.; Rodriguez, F.; Hachem, R.; Fisher, C.E.; Limaye, A.P.; Omar, A.; et al. Respiratory viral infection in lung transplantation induces exosomes that trigger chronic rejection. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 2020, 39, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Sadeghipour, S.; Mathias, R.A. Herpesviruses hijack host exosomes for viral pathogenesis. Semin. Cell Dev. Biol. 2017, 67, 91–100. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, Q.; Xie, Y.; Zuo, L.; Zhu, F.; Lu, J. Extracellular vesicles: Novel vehicles in herpesvirus infection. Virol. Sin. 2017, 32, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Wald, A.; Corey, L. Persistence in the population: Epidemiology, transmission. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Roizman, B.; Zhou, G.; Du, T. Checkpoints in productive and latent infections with herpes simplex virus 1: Conceptualization of the issues. J. Neurovirol. 2011, 17, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Held, K.; Derfuss, T. Control of HSV-1 latency in human trigeminal ganglia—Current overview. J. Neurovirol. 2011, 17, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.; Vanicek, J.; Robins, H.; Shenk, T.; Levine, A.J. Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: Implications for latency. Proc. Natl. Acad. Sci. USA 2008, 105, 5453–5458. [Google Scholar] [CrossRef] [Green Version]
- Obara, Y.; Furuta, Y.; Takasu, T.; Suzuki, S.; Suzuki, H.; Matsukawa, S.; Fujioka, Y.; Takahashi, H.; Kurata, T.; Nagashima, K. Distribution of herpes simplex virus types 1 and 2 genomes in human spinal ganglia studied by PCR and in situ hybridization. J. Med. Virol. 1997, 52, 136–142. [Google Scholar] [CrossRef]
- Kennedy, P.G.; Chaudhuri, A. Herpes simplex encephalitis. J. Neurol. Neurosurg. Psychiatry 2002, 73, 237–238. [Google Scholar] [CrossRef]
- Whitley, R.J. Herpes simplex encephalitis: Adolescents and adults. Antivir. Res. 2006, 71, 141–148. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Bellamy, A.R.; Hook, E.W., 3rd; Levin, M.J.; Wald, A.; Ewell, M.G.; Wolff, P.A.; Deal, C.D.; Heineman, T.C.; Dubin, G.; et al. Epidemiology, clinical presentation, and antibody response to primary infection with herpes simplex virus type 1 and type 2 in young women. Clin. Infect. Dis. 2013, 56, 344–351. [Google Scholar] [CrossRef]
- Karasneh, G.A.; Shukla, D. Herpes simplex virus infects most cell types in vitro: Clues to its success. Virol. J. 2011, 8, 481. [Google Scholar] [CrossRef] [Green Version]
- Agelidis, A.M.; Shukla, D. Cell entry mechanisms of HSV: What we have learned in recent years. Future Virol. 2015, 10, 1145–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldwein, E.E.; Krummenacher, C. Entry of herpesviruses into mammalian cells. Cell Mol. Life Sci 2008, 65, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Reske, A.; Pollara, G.; Krummenacher, C.; Chain, B.M.; Katz, D.R. Understanding HSV-1 entry glycoproteins. Rev. Med. Virol. 2007, 17, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, J.; Shukla, D. Viral entry mechanisms: Cellular and viral mediators of herpes simplex virus entry. Febs J. 2009, 276, 7228–7236. [Google Scholar] [CrossRef]
- Shukla, D.; Spear, P.G. Herpesviruses and heparan sulfate: An intimate relationship in aid of viral entry. J. Clin. Investig. 2001, 108, 503–510. [Google Scholar] [CrossRef]
- Schweighardt, B.; Atwood, W.J. Virus receptors in the human central nervous system. J. Neurovirol. 2001, 7, 187–195. [Google Scholar]
- Hilterbrand, A.T.; Heldwein, E.E. Go go gadget glycoprotein!: HSV-1 draws on its sizeable glycoprotein tool kit to customize its diverse entry routes. PLoS Pathog. 2019, 15, e1007660. [Google Scholar] [CrossRef]
- Owen, D.J.; Crump, C.M.; Graham, S.C. Tegument Assembly and Secondary Envelopment of Alphaherpesviruses. Viruses 2015, 7, 5084–5114. [Google Scholar] [CrossRef] [Green Version]
- Calistri, A.; Sette, P.; Salata, C.; Cancellotti, E.; Forghieri, C.; Comin, A.; Gottlinger, H.; Campadelli-Fiume, G.; Palu, G.; Parolin, C. Intracellular trafficking and maturation of herpes simplex virus type 1 gB and virus egress require functional biogenesis of multivesicular bodies. J. Virol. 2007, 81, 11468–11478. [Google Scholar] [CrossRef] [Green Version]
- Crump, C.M.; Yates, C.; Minson, T. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J. Virol. 2007, 81, 7380–7387. [Google Scholar] [CrossRef] [Green Version]
- Campadelli-Fiume, G. The egress of alphaherpesviruses from the cell. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Satoh, T.; Arii, J.; Suenaga, T.; Wang, J.; Kogure, A.; Uehori, J.; Arase, N.; Shiratori, I.; Tanaka, S.; Kawaguchi, Y.; et al. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 2008, 132, 935–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suenaga, T.; Satoh, T.; Somboonthum, P.; Kawaguchi, Y.; Mori, Y.; Arase, H. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc. Natl. Acad. Sci. USA 2010, 107, 866–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szilagyi, J.F.; Cunningham, C. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J. Gen. Virol. 1991, 72 Pt 3, 661–668. [Google Scholar] [CrossRef] [PubMed]
- McLauchlan, J.; Addison, C.; Craigie, M.C.; Rixon, F.J. Noninfectious L-particles supply functions which can facilitate infection by HSV-1. Virology 1992, 190, 682–688. [Google Scholar] [CrossRef]
- Heilingloh, C.S.; Krawczyk, A. Role of L-Particles during Herpes Simplex Virus Infection. Front. Microbiol. 2017, 8, 2565. [Google Scholar] [CrossRef] [Green Version]
- Kalamvoki, M.; Deschamps, T. Extracellular vesicles during Herpes Simplex Virus type 1 infection: An inquire. Virol. J. 2016, 13, 63. [Google Scholar] [CrossRef] [Green Version]
- Heilingloh, C.S.; Kummer, M.; Muhl-Zurbes, P.; Drassner, C.; Daniel, C.; Klewer, M.; Steinkasserer, A. L Particles Transmit Viral Proteins from Herpes Simplex Virus 1-Infected Mature Dendritic Cells to Uninfected Bystander Cells, Inducing CD83 Downmodulation. J. Virol. 2015, 89, 11046–11055. [Google Scholar] [CrossRef] [Green Version]
- Dargan, D.J.; Patel, A.H.; Subak-Sharpe, J.H. PREPs: Herpes simplex virus type 1-specific particles produced by infected cells when viral DNA replication is blocked. J. Virol. 1995, 69, 4924–4932. [Google Scholar] [CrossRef] [Green Version]
- Temme, S.; Eis-Hubinger, A.M.; McLellan, A.D.; Koch, N. The herpes simplex virus-1 encoded glycoprotein B diverts HLA-DR into the exosome pathway. J. Immunol. 2010, 184, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Bello-Morales, R.; Praena, B.; de la Nuez, C.; Rejas, M.T.; Guerra, M.; Galan-Ganga, M.; Izquierdo, M.; Calvo, V.; Krummenacher, C.; Lopez-Guerrero, J.A. Role of Microvesicles in the Spread of Herpes Simplex Virus 1 in Oligodendrocytic Cells. J. Virol. 2018, 92, e00088-18. [Google Scholar] [CrossRef] [Green Version]
- Kalamvoki, M.; Du, T.; Roizman, B. Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing STING, viral mRNAs, and microRNAs. Proc. Natl. Acad. Sci. USA 2014, 111, E4991–E4996. [Google Scholar] [CrossRef] [Green Version]
- Deschamps, T.; Kalamvoki, M. Extracellular Vesicles Released by Herpes Simplex Virus 1-Infected Cells Block Virus Replication in Recipient Cells in a STING-Dependent Manner. J. Virol. 2018, 92, e01102-18. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12 (Suppl. 1–13), 19–30. [Google Scholar] [CrossRef] [Green Version]
- Gerber, P.P.; Cabrini, M.; Jancic, C.; Paoletti, L.; Banchio, C.; von Bilderling, C.; Sigaut, L.; Pietrasanta, L.I.; Duette, G.; Freed, E.O.; et al. Rab27a controls HIV-1 assembly by regulating plasma membrane levels of phosphatidylinositol 4,5-bisphosphate. J. Cell Biol. 2015, 209, 435–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraile-Ramos, A.; Cepeda, V.; Elstak, E.; van der Sluijs, P. Rab27a is required for human cytomegalovirus assembly. PLoS ONE 2010, 5, e15318. [Google Scholar] [CrossRef] [PubMed]
- Bello-Morales, R.; Crespillo, A.J.; Fraile-Ramos, A.; Tabares, E.; Alcina, A.; Lopez-Guerrero, J.A. Role of the small GTPase Rab27a during herpes simplex virus infection of oligodendrocytic cells. BMC Microbiol. 2012, 12, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bello-Morales, R.; Lopez-Guerrero, J.A. Extracellular Vesicles in Herpes Viral Spread and Immune Evasion. Front. Microbiol. 2018, 9, 2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabowska, K.; Wachalska, M.; Graul, M.; Rychlowski, M.; Bienkowska-Szewczyk, K.; Lipinska, A.D. Alphaherpesvirus gB Homologs Are Targeted to Extracellular Vesicles, but They Differentially Affect MHC Class II Molecules. Viruses 2020, 12, 429. [Google Scholar] [CrossRef] [Green Version]
- Gershon, M.; Gershon, A. Varicella-Zoster Virus and the Enteric Nervous System. J. Infect. Dis. 2018, 218 (Suppl. 2), S113–S119. [Google Scholar] [CrossRef]
- Mori, Y.; Koike, M.; Moriishi, E.; Kawabata, A.; Tang, H.; Oyaizu, H.; Uchiyama, Y.; Yamanishi, K. Human herpesvirus-6 induces MVB formation, and virus egress occurs by an exosomal release pathway. Traffic 2008, 9, 1728–1742. [Google Scholar] [CrossRef] [Green Version]
- Ota, M.; Serada, S.; Naka, T.; Mori, Y. MHC class I molecules are incorporated into human herpesvirus-6 viral particles and released into the extracellular environment. Microbiol. Immunol. 2014, 58, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraile-Ramos, A.; Pelchen-Matthews, A.; Risco, C.; Rejas, M.T.; Emery, V.C.; Hassan-Walker, A.F.; Esteban, M.; Marsh, M. The ESCRT machinery is not required for human cytomegalovirus envelopment. Cell. Microbiol. 2007, 9, 2955–2967. [Google Scholar] [CrossRef] [PubMed]
- Schauflinger, M.; Fischer, D.; Schreiber, A.; Chevillotte, M.; Walther, P.; Mertens, T.; von Einem, J. The tegument protein UL71 of human cytomegalovirus is involved in late envelopment and affects multivesicular bodies. J. Virol. 2011, 85, 3821–3832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zicari, S.; Arakelyan, A.; Palomino, R.A.N.; Fitzgerald, W.; Vanpouille, C.; Lebedeva, A.; Schmitt, A.; Bomsel, M.; Britt, W.; Margolis, L. Human cytomegalovirus-infected cells release extracellular vesicles that carry viral surface proteins. Virology 2018, 524, 97–105. [Google Scholar] [CrossRef]
- Walker, J.D.; Maier, C.L.; Pober, J.S. Cytomegalovirus-infected human endothelial cells can stimulate allogeneic CD4+ memory T cells by releasing antigenic exosomes. J. Immunol. 2009, 182, 1548–1559. [Google Scholar] [CrossRef] [Green Version]
- Meckes, D.G., Jr.; Gunawardena, H.P.; Dekroon, R.M.; Heaton, P.R.; Edwards, R.H.; Ozgur, S.; Griffith, J.D.; Damania, B.; Raab-Traub, N. Modulation of B-cell exosome proteins by gamma herpesvirus infection. Proc. Natl. Acad. Sci. USA 2013, 110, E2925–E2933. [Google Scholar] [CrossRef] [Green Version]
- Vazirabadi, G.; Geiger, T.R.; Coffin, W.F., 3rd; Martin, J.M. Epstein-Barr virus latent membrane protein-1 (LMP-1) and lytic LMP-1 localization in plasma membrane-derived extracellular vesicles and intracellular virions. J. Gen. Virol. 2003, 84 Pt 8, 1997–2008. [Google Scholar] [CrossRef]
- Meckes, D.G., Jr.; Shair, K.H.; Marquitz, A.R.; Kung, C.P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef] [Green Version]
- Cone, A.S.; York, S.B.; Meckes, D.G., Jr. Extracellular Vesicles in Epstein-Barr Virus Pathogenesis. Curr. Clin. Microbiol. Rep. 2019, 6, 121–131. [Google Scholar] [CrossRef]
- Kieser, A.; Sterz, K.R. The Latent Membrane Protein 1 (LMP1). Curr. Top. Microbiol. Immunol. 2015, 391, 119–149. [Google Scholar]
- Liao, C.; Zhou, Q.; Zhang, Z.; Wu, X.; Zhou, Z.; Li, B.; Peng, J.; Shen, L.; Li, D.; Luo, X.; et al. Epstein-Barr virus-encoded latent membrane protein 1 promotes extracellular vesicle secretion through syndecan-2 and synaptotagmin-like-4 in nasopharyngeal carcinoma cells. Cancer Sci. 2020, 111, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.; Middeldorp, J.; Sculley, T. Localization of the Epstein-Barr virus protein LMP 1 to exosomes. J. Gen. Virol. 2003, 84 Pt 7, 1871–1879. [Google Scholar] [CrossRef] [PubMed]
- Verweij, F.J.; van Eijndhoven, M.A.; Hopmans, E.S.; Vendrig, T.; Wurdinger, T.; Cahir-McFarland, E.; Kieff, E.; Geerts, D.; van der Kant, R.; Neefjes, J.; et al. LMP1 association with CD63 in endosomes and secretion via exosomes limits constitutive NF-kappaB activation. EMBO J. 2011, 30, 2115–2129. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, S.N.; Nkosi, D.; Conlon, M.M.; York, S.B.; Liu, X.; Tremblay, D.C.; Meckes, D.G., Jr. CD63 Regulates Epstein-Barr Virus LMP1 Exosomal Packaging, Enhancement of Vesicle Production, and Noncanonical NF-kappaB Signaling. J. Virol. 2017, 91, e02251-16. [Google Scholar] [CrossRef] [Green Version]
- Nanbo, A.; Kawanishi, E.; Yoshida, R.; Yoshiyama, H. Exosomes derived from Epstein-Barr virus-infected cells are internalized via caveola-dependent endocytosis and promote phenotypic modulation in target cells. J. Virol. 2013, 87, 10334–10347. [Google Scholar] [CrossRef] [Green Version]
- Pegtel, D.M.; Cosmopoulos, K.; Thorley-Lawson, D.A.; van Eijndhoven, M.A.; Hopmans, E.S.; Lindenberg, J.L.; de Gruijl, T.D.; Wurdinger, T.; Middeldorp, J.M. Functional delivery of viral miRNAs via exosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 6328–6333. [Google Scholar] [CrossRef] [Green Version]
- Gourzones, C.; Gelin, A.; Bombik, I.; Klibi, J.; Verillaud, B.; Guigay, J.; Lang, P.; Temam, S.; Schneider, V.; Amiel, C.; et al. Extra-cellular release and blood diffusion of BART viral micro-RNAs produced by EBV-infected nasopharyngeal carcinoma cells. Virol. J. 2010, 7, 271. [Google Scholar] [CrossRef] [Green Version]
- Piedade, D.; Azevedo-Pereira, J.M. The Role of microRNAs in the Pathogenesis of Herpesvirus Infection. Viruses 2016, 8, 156. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, A.R.; Shango, J.; Seal, A.; Shukla, D.; Nares, S. Viral miRNAs Alter Host Cell miRNA Profiles and Modulate Innate Immune Responses. Front. Immunol. 2018, 9, 433. [Google Scholar] [CrossRef]
- Li, C.; He, H.; Wang, J.; Xia, X.; Zhang, M.; Wu, Q. Possible roles of exosomal miRNAs in the pathogenesis of oral lichen planus. Am. J. Transl. Res. 2019, 11, 5313–5323. [Google Scholar]
- Ding, M.; Wang, X.; Wang, C.; Liu, X.; Zen, K.; Wang, W.; Zhang, C.Y.; Zhang, C. Distinct expression profile of HCMV encoded miRNAs in plasma from oral lichen planus patients. J. Transl. Med. 2017, 15, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Nanbo, A.; Sun, L.; Lin, Z. Extracellular Vesicles in Epstein-Barr Virus’ Life Cycle and Pathogenesis. Microorganisms 2019, 7, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, J.A.; Tomita, Y.; Jankowska-Gan, E.; Lema, D.A.; Arvedson, M.P.; Nair, A.; Bracamonte-Baran, W.; Zhou, Y.; Meyer, K.K.; Zhong, W.; et al. Treg-Cell-Derived IL-35-Coated Extracellular Vesicles Promote Infectious Tolerance. Cell Rep. 2020, 30, 1039–1051.e5. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tsai, M.H.; Shumilov, A.; Baccianti, F.; Tsao, S.W.; Poirey, R.; Delecluse, H.J. Epstein-Barr virus ncRNA from a nasopharyngeal carcinoma induces an inflammatory response that promotes virus production. Nat. Microbiol. 2019, 4, 2475–2486. [Google Scholar] [CrossRef]
- Zheng, J.; Shi, Y.; Feng, Z.; Zheng, Y.; Li, Z.; Zhao, Y.; Wang, Y. Oncogenic effects of exosomes in gamma-herpesvirus-associated neoplasms. J. Cell. Physiol. 2019, 234, 19167–19179. [Google Scholar] [CrossRef] [PubMed]
- Chugh, P.E.; Sin, S.H.; Ozgur, S.; Henry, D.H.; Menezes, P.; Griffith, J.; Eron, J.J.; Damania, B.; Dittmer, D.P. Systemically circulating viral and tumor-derived microRNAs in KSHV-associated malignancies. PLoS Pathog. 2013, 9, e1003484. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Feng, Z.; Yuan, G.; Emerson, C.C.; Stewart, P.L.; Ye, F.; Jin, G. Human Immunodeficiency Virus-Associated Exosomes Promote Kaposi’s Sarcoma-Associated Herpesvirus Infection via the Epidermal Growth Factor Receptor. J. Virol. 2020, 94, e01782-19. [Google Scholar] [CrossRef] [Green Version]
- Alonso, M.A.; Weissman, S.M. cDNA cloning and sequence of MAL, a hydrophobic protein associated with human T-cell differentiation. Proc. Natl. Acad. Sci. USA 1987, 84, 1997–2001. [Google Scholar] [CrossRef] [Green Version]
- Millan, J.; Puertollano, R.; Fan, L.; Rancano, C.; Alonso, M.A. The MAL proteolipid is a component of the detergent-insoluble membrane subdomains of human T-lymphocytes. Biochem. J. 1997, 321 Pt 1, 247–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, P.; Puertollano, R.; Alonso, M.A. Structural and biochemical similarities reveal a family of proteins related to the MAL proteolipid, a component of detergent-insoluble membrane microdomains. Biochem. Biophys. Res. Commun. 1997, 232, 618–621. [Google Scholar] [CrossRef]
- Marazuela, M.; Acevedo, A.; Adrados, M.; Garcia-Lopez, M.A.; Alonso, M.A. Expression of MAL, an integral protein component of the machinery for raft-mediated pical transport, in human epithelia. J. Histochem. Cytochem. 2003, 51, 665–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, M. MAL, a proteolipid in glycosphingolipid enriched domains: Functional implications in myelin and beyond. Prog. Neurobiol. 2000, 60, 531–544. [Google Scholar] [CrossRef]
- Bijlard, M.; de Jonge, J.C.; Klunder, B.; Nomden, A.; Hoekstra, D.; Baron, W. MAL Is a Regulator of the Recruitment of Myelin Protein PLP to Membrane Microdomains. PLoS ONE 2016, 11, e0155317. [Google Scholar] [CrossRef] [PubMed]
- Puertollano, R.; Martin-Belmonte, F.; Millan, J.; de Marco, M.C.; Albar, J.P.; Kremer, L.; Alonso, M.A. The MAL proteolipid is necessary for normal apical transport and accurate sorting of the influenza virus hemagglutinin in Madin-Darby canine kidney cells. J. Cell Biol. 1999, 145, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Martin-Belmonte, F.; Arvan, P.; Alonso, M.A. MAL mediates apical transport of secretory proteins in polarized epithelial Madin-Darby canine kidney cells. J. Biol. Chem. 2001, 276, 49337–49342. [Google Scholar] [CrossRef] [Green Version]
- Ventimiglia, L.N.; Fernandez-Martin, L.; Martinez-Alonso, E.; Anton, O.M.; Guerra, M.; Martinez-Menarguez, J.A.; Andres, G.; Alonso, M.A. Cutting Edge: Regulation of Exosome Secretion by the Integral MAL Protein in T Cells. J. Immunol. 2015, 195, 810–814. [Google Scholar] [CrossRef] [Green Version]
- Ventimiglia, L.N.; Alonso, M.A. Biogenesis and Function of T Cell-Derived Exosomes. Front. Cell Dev. Biol. 2016, 4, 84. [Google Scholar] [CrossRef] [Green Version]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Chazal, N.; Gerlier, D. Virus entry, assembly, budding, and membrane rafts. Microbiol. Mol. Biol. Rev. Mmbr 2003, 67, 226–237. [Google Scholar] [CrossRef] [Green Version]
- Nayak, D.P.; Barman, S. Role of lipid rafts in virus assembly and budding. Adv. Virus Res. 2002, 58, 1–28. [Google Scholar]
- Bukrinsky, M.I.; Mukhamedova, N.; Sviridov, D. Lipid rafts and pathogens: The art of deception and exploitation. J. Lipid Res. 2020, 61, 601–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hantak, M.P.; Qing, E.; Earnest, J.T.; Gallagher, T. Tetraspanins: Architects of Viral Entry and Exit Platforms. J. Virol. 2019, 93, e01429-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Guerrero, J.A.; de la Nuez, C.; Praena, B.; Sanchez-Leon, E.; Krummenacher, C.; Bello-Morales, R. Herpes Simplex Virus 1 Spread in Oligodendrocytic Cells Is Highly Dependent on MAL Proteolipid. J. Virol. 2020, 94, e01739-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bello-Morales, R.; Ripa, I.; López-Guerrero, J.A. Extracellular Vesicles in Viral Spread and Antiviral Response. Viruses 2020, 12, 623. https://doi.org/10.3390/v12060623
Bello-Morales R, Ripa I, López-Guerrero JA. Extracellular Vesicles in Viral Spread and Antiviral Response. Viruses. 2020; 12(6):623. https://doi.org/10.3390/v12060623
Chicago/Turabian StyleBello-Morales, Raquel, Inés Ripa, and José Antonio López-Guerrero. 2020. "Extracellular Vesicles in Viral Spread and Antiviral Response" Viruses 12, no. 6: 623. https://doi.org/10.3390/v12060623
APA StyleBello-Morales, R., Ripa, I., & López-Guerrero, J. A. (2020). Extracellular Vesicles in Viral Spread and Antiviral Response. Viruses, 12(6), 623. https://doi.org/10.3390/v12060623