1. Introduction
Programmed death-ligand 1 (PD-L1) is highly expressed in diverse cancers, and is regarded as a major contributor to the immune incompetency of immune cells in cancer microenvironments through the ligation between PD-L1 and programmed cell death protein 1 (PD-1) on T cells that generate strong suppressive signals in the activated T cell to make it anergic [
1,
2,
3,
4,
5]. After the reports in the
New England Journal of Medicine in June 2012 on the phase I studies of anti-PD-L1 and PD-1 antibodies amazed the whole oncology field [
6,
7], immune checkpoint inhibitory antibodies have become some of the most promising antibody drugs to treat several cancers in the clinic [
8]. The reinvigoration of immune reactivity by PD-1 and PD-L1 targeting antibodies toward several cancers has been quite successful; therefore, the enhanced usage of these antibodies as a “backbone” regimen of anticancer combination treatments is likely [
9].
“Phage display” was initially described by Smith in 1985 [
10], in which the coating proteins of filamentous bacteriophages fused with exogenous peptides were expressed on their surface and used for the enrichment of specific phages. Subsequently, Adalimumab (Humira) was developed in 1990 by Winter as the first phage-display-derived therapeutic antibody to neutralize tumor necrosis factor-alpha (TNF-α) for immunological disease treatment. Antibody phage-display technologies were further developed by several other groups (Winter and McCafferty, Cambridge, UK; Lerner and Barbas, California, USA; and Breitling and Dübel, Heidelberg, Germany) [
11,
12,
13,
14,
15,
16]. In 2002, Adalimumab (Humira) was approved by the FDA for the treatment of rheumatoid arthritis and is currently the best-selling drug in the world [
17]. In 2018, Smith and Winter were jointly awarded the Nobel Prize in chemistry for their contributions to the development of the phage-display technology.
Phage display became the most powerful and commonly used technology for antibody drug development and has been used in various other research areas, including enzyme optimization, affinity maturation of antibodies, the development of nanovehicles (nanomedicines), epitope mapping, vaccine development, and biomolecular interaction studies [
18,
19,
20,
21,
22,
23,
24]. For antibody screening using phage display, a single-pot antibody library is applied to target antigens and 3‒4 rounds of panning are usually performed to enrich the antibody binders. At the end of panning, bacterial colonies infected with individual phages are used for the characterization of each clone’s binding to an antigen. Usually, phage particles or scFv/Fab are used for the first binding assessment of each antibody without detailed normalization information [
25]; the antibody conversion to IgG format commonly follows. These IgG-format purified antibodies are applied to most antibody characterizations, such as enzyme-linked immunosorbent assay (ELISA), fluorescence-activated cell sorting (FACS) analysis on cell surface antigens, direct protein interaction analysis through surface plasmon resonance (SPR), and other in vitro cell-based assays, through which functional antibody candidates can usually be selected.
However, this typical phage-display process requires a lot of effort and resources to get the functional antibody candidates in the end; IgG conversion and antibody production and purification from a mammalian cell system in large quantities, especially, are difficult steps for academic researchers [
26,
27]. Therefore, the elimination of unqualified antibody candidates at an earlier stage and earlier functional characterization of antibody clones will speed up the selection of highly functional candidates and result in more successful therapeutic antibody development.
Biolayer interferometry (BLI) is used to characterize direct antibody‒antigen interactions, especially in the Octet system from Pall ForteBio (Fremont, CA, USA) [
28]. BLI is a label-free technique that enables real-time monitoring of biomolecule interactions by analyzing optical interference pattern changes on protein-coated sensor chip surfaces generated by mass increases from analyte‒protein interactions. Its mechanism, basically the same as the surface plasmon resonance effect, is taken into account by the well-known Biacore analytical system from Cytiva (Marlborough, MA, USA). However, 96-well or 384-well BLI assay facilitates high-throughput analysis, which offers greater potential in drug development applications.
In this study, we designed a new BLI-based functional scFv screening method and applied it to screen out functional antibodies targeting the PD-L1 immune-checkpoint ligand protein. The BLI assay in an earlier stage showed its high functional relevance with an in vitro cell-based assay and that extended well to an in vivo syngeneic mouse model study. Our study showed that the BLI-based assay is highly beneficial for functional antibody screening and drug candidate development.
2. Materials and Methods
2.1. Materials and Reagents
Human and mouse PD-L1 cDNA were purchased from Sino Biologicals, Inc. (Beijing, China). PCR was performed using the AccuPrime pfx DNA polymerase (Invitrogen, Carlsbad, CA, USA). All restriction enzymes and T4 ligase were purchased from New England Biolabs (Ipswich, MA, USA). Recombinant mouse interferon gamma (IFN-γ) was purchased from Pepro Tech (Rocky Hill, NJ, USA). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), ethanolamine, and Amine Reactive Second-Generation (AR2G) biosensors were obtained from ForteBio (Fremont, CA, USA).
2.2. Cell Lines and Transfection
PC3 and 293T cells were obtained from the American Type Culture Collection (Manassas, VA, USA). The murine colon cancer cell line MC38 was a kind gift from Dr. Sin, Kangwon National University (Chuncheon, Korea). Cells were cultured in RPMI-1640 or DMEM (Gibco, Grand Island, NY, USA), supplemented with 10% v/v fetal bovine serum (FBS) and 1% v/v penicillin/streptomycin (Gibco). FreeStyle 293F cells were maintained in Freestyle 293 expression medium (Invitrogen). The 293T and FreeStyle 293F cells were transfected using Lipofectamine 2000 (Invitrogen) or FectoPRO transfection reagent (Polyplus-transfection, Illkirch, France).
2.3. Phage Panning
For our phage-display antibody screening, a human naïve antibody library kindly provided by LG Life Sciences, a private biotech company in South Korea, through a material transfer agreement was used to isolate human antibody clones binding to human PD-L1. The antibody library has about 1.2 × 10
11 diversity and each scFv is expressed with a c-Myc tag at its C-terminus. The human naïve scFv library was re-amplified as scFv-displaying phages and the phage pools from each round of panning were generated using ER2738 cells (New England Biolabs). Phage-display experiments including library reamplification and phage panning were mainly performed with the protocols described in
Phage Display: A Laboratory Manual [
29], with the following additional details.
To rescue scFv-displaying phages, frozen library stocks were thawed and inoculated into super broth (SB) medium (containing 2% glucose, 35 μg/mL chloramphenicol). A VCSM13 helper phage (1012 colony-forming units) was then added to the library cells in the exponential growth phase. After overnight culture, the phage was purified by PEG precipitation (final 5% w/v polyethylene glycol 8000 (PEG8000), 0.5 M NaCl).
Biopanning for the enrichment of scFv binders was performed using human IgG (12.5 μg/mL, Green Cross Corp., Yong-in, Korea)-coated immunotubes (Nunc, Roskilde, Denmark) or PD-L1-conjugated magnetic beads (4 μg/107 beads, Dynabeads M270 epoxy, Invitrogen, Carlsbad, CA, USA). In order to remove nonspecific scFv phage binders from the Fc region, a subtraction reaction was performed using a human IgG-coated immunotube for 2 h at room temperature, then the scFv phages in the supernatant were incubated for 1 h at 4 °C with PD-L1-conjugated magnetic beads. The phage-PD-L1 antigens were washed one time for the first panning, 2‒3 times for the second and third panning, and 3‒5 times for the fourth panning with PBS (containing 0.1% Tween 20) to introduce increased stringency and obtain high-affinity binders. Bound phages were eluted with 0.25% trypsin (Gibco). Eluted phages were amplified by infection to E. coli ER2738 and the phage rescued as outline above. A total of four rounds of panning were performed to enrich the scFv antibody binders and individual colonies picked randomly from the third and fourth rounds of panning were sought after the target binding assays.
2.4. scFv Expression and TES Periplasmic Extraction
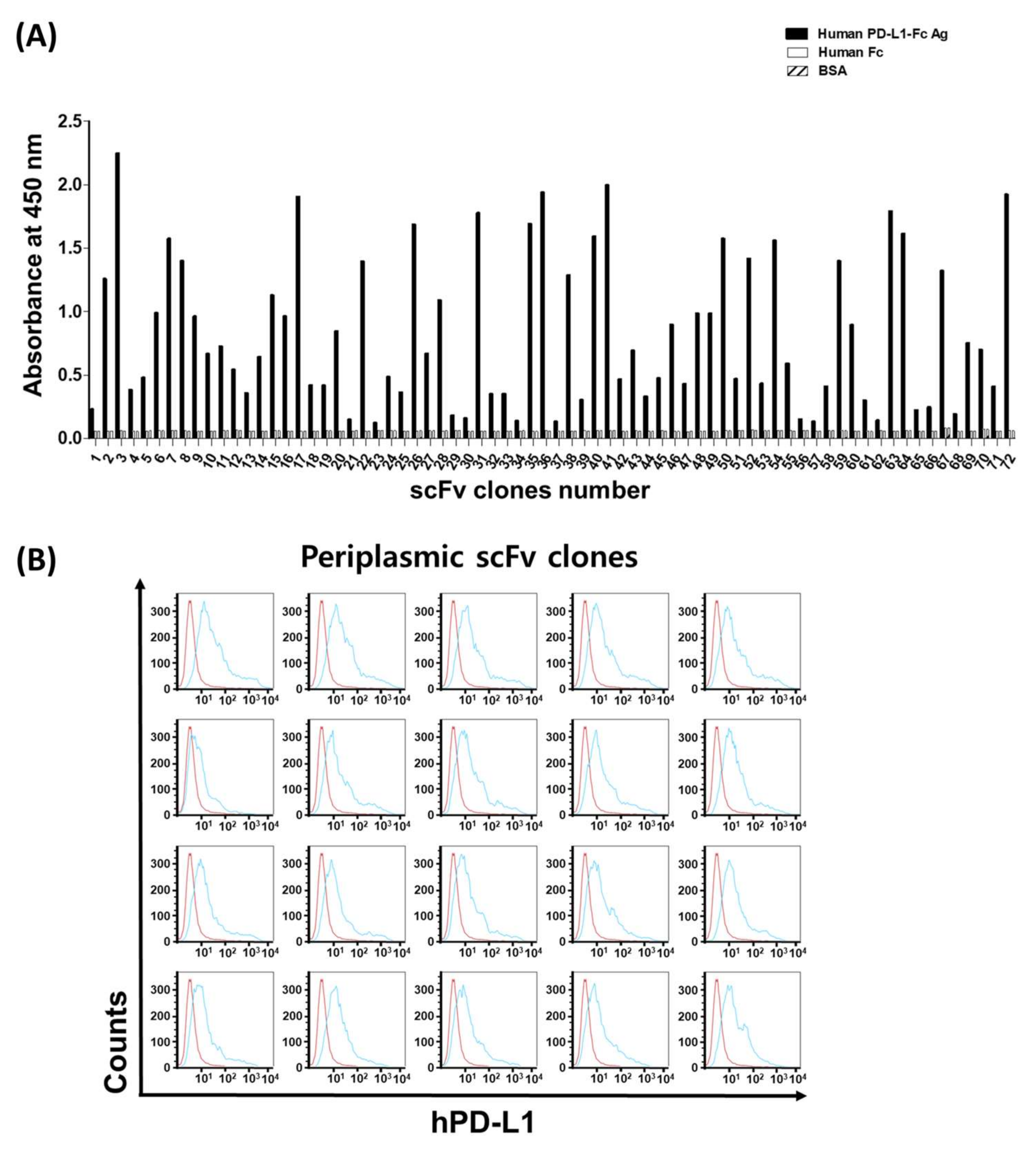
Single colonies expressing individual scFvs from the third and fourth panning pools were randomly picked and grown overnight in a 96-well plate containing 300 μL/well of super broth (SB) with carbenicillin. Twenty microliters of overnight-grown cultures were inoculated in 900 μL/well of SB medium carbenicillin and cultured at 30 °C with shaking. When the absorbance at 600 nm reached 0.7, IPTG was added to each well (1 mM final concentration) and cells were grown overnight at 30 °C with shaking. After tabletop centrifugation, a periplasmic extract was obtained using the osmotic shock method. Briefly, after centrifugation at 3500 rpm at room temperature (to discard the culture media), the cell pellets were resuspended in 320 μL of ice-cold extraction solution (TES buffer, containing 20% w/v sucrose, 50 mM Tris, 1 mM EDTA, pH 8.0) and after 5 min on ice bath, 480 μL of ice-cold 0.2 × TES buffer was added to each well. After 30 min incubation on ice, the suspension was centrifuged at 3500 rpm, 4 °C, and scFv-containing periplasmic crude extracts were obtained as the supernatants. scFv-containing periplasmic crude extracts were used for the enzyme-linked immunosorbent assay (ELISA), flow cytometry, and biolayer interferometry (BLI)-based assay.
2.5. Expression of IgG Format Antibodies Using a Two-Vector System for Heavy and Light
With germline sequence information for 72 scFv clones, the primers for those VH and VL domains were synthesized and used for PCR amplification of individual VH and VL fragments (
Table S1). Each VH primer was designed to have restriction sites for KpnI or BamHI at the 5′ ends and NheI at the 3′ ends. For the VL primers, BamHI and BsiWI restriction sites were incorporated at the 5′ and 3′ ends, respectively. Amplified VH and VL fragments of each clone were ligated into the pCEP4 mammalian expression vector (Invitrogen) as a fused form, either with CH1-hinge-CH2-CH3 for the heavy chain expression or with C
κ for the light chain expression. Matching heavy and light chain vectors for each clone were co-transfected into FreeStyle 293-F cells (Thermo Fisher) with a 2:1 (light vs. heavy) DNA ratio using FectoPRO transfection reagent (Polyplus-transfection) as per the manufacturer’s instructions; after 12 days, the culture medium was harvested. The IgG antibodies were purified by open-column chromatography using protein A agarose beads (GenScript, Piscataway, NJ, USA).
After dialysis with phosphate-buffered saline at pH 7.4 (PBS), the antibody concentration was quantified using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and the purity of each antibody was evaluated by SDS/polyacrylamide gel electrophoresis (PAGE) and Coomassie brilliant blue staining.
2.6. ELISA
The specificity and binding activity of individual scFv and IgG were assessed by ELISA. The human PD-L1-Fc and human IgG were applied to a 96-well ELISA plate at a concentration of 10 µg/mL in PBS at 4 °C overnight. After being washed three times with PBS containing 0.05% v/v Tween 20 (PBST), the wells were blocked with 3% w/v BSA in PBST for 1 h at 37 °C. Then, 25 μL/well scFv-containing periplasmic crude extracts or 100 ng/well purified IgGs were added and incubated for 1 h at 37 °C. After washing, for scFv ELISA, 2 µg/mL mouse anti-c-Myc IgG (Sigma-Aldrich, St. Louis, MO, USA) was added for 1 h at 37 °C, and, after washing, horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (1/10,000, Jackson ImmunoResearch, West Grove, PA, USA) in PBST containing 3% BSA was incubated for 1 h at 37 °C. For ELISA using IgG, HRP-conjugated goat anti-human kappa light chain IgG (1:5000, Sigma) was added for 1 h. For detection, 100 μL/well of 3,3’,5,5’-Tetramethyl–benzidine (TMB) substrate (BD Biosciences, San Jose, CA, USA) was used and the absorbance read at 450 nm using a microtiter plate reader (VICTOR X4, Perkin Elmer, Waltham, MA, USA).
2.7. Flow Cytometry
The specific binding of the scFv-containing periplasmic crude extracts and purified IgGs directed to cell-surface human PD-L1 and mouse PD-L1 was analyzed by BD FACSCalibur (BD Biosciences). About 5 × 105 cells were used in each experiment. Harvested cells were resuspended in 100 μL FACS buffer (2% BSA and 0.02% NaN3 in PBS) and then mixed with 100 μL scFv-containing periplasmic crude extracts or 10 µg/mL purified IgG in FACS buffer on ice for 1 h. After washing with FACS buffer, cells were treated for 1 h with 2 μg/mL mouse anti-c-Myc antibody and 1 h with Alexa Fluor 488-conjugated goat anti-mouse IgG (1:400, Jackson ImmunoResearch) for periplasmic scFv staining. For flow cytometry using IgG, after 1 h incubation with each IgG clones, Alexa Fluor 488-conjugated goat anti-human IgG (1:400, Jackson ImmunoResearch) was treated for 1 h. Cells were washed between the reactions with a FACS buffer. All samples were measured by a FACSCalibur flow cytometer and CELL QUEST program (BD Biosciences), and the data were analyzed by FlowJo software (FlowJo LLC, Ashland, OR, USA).
2.8. BLI-Based PD-1/PD-L1 Binding Inhibition Assay
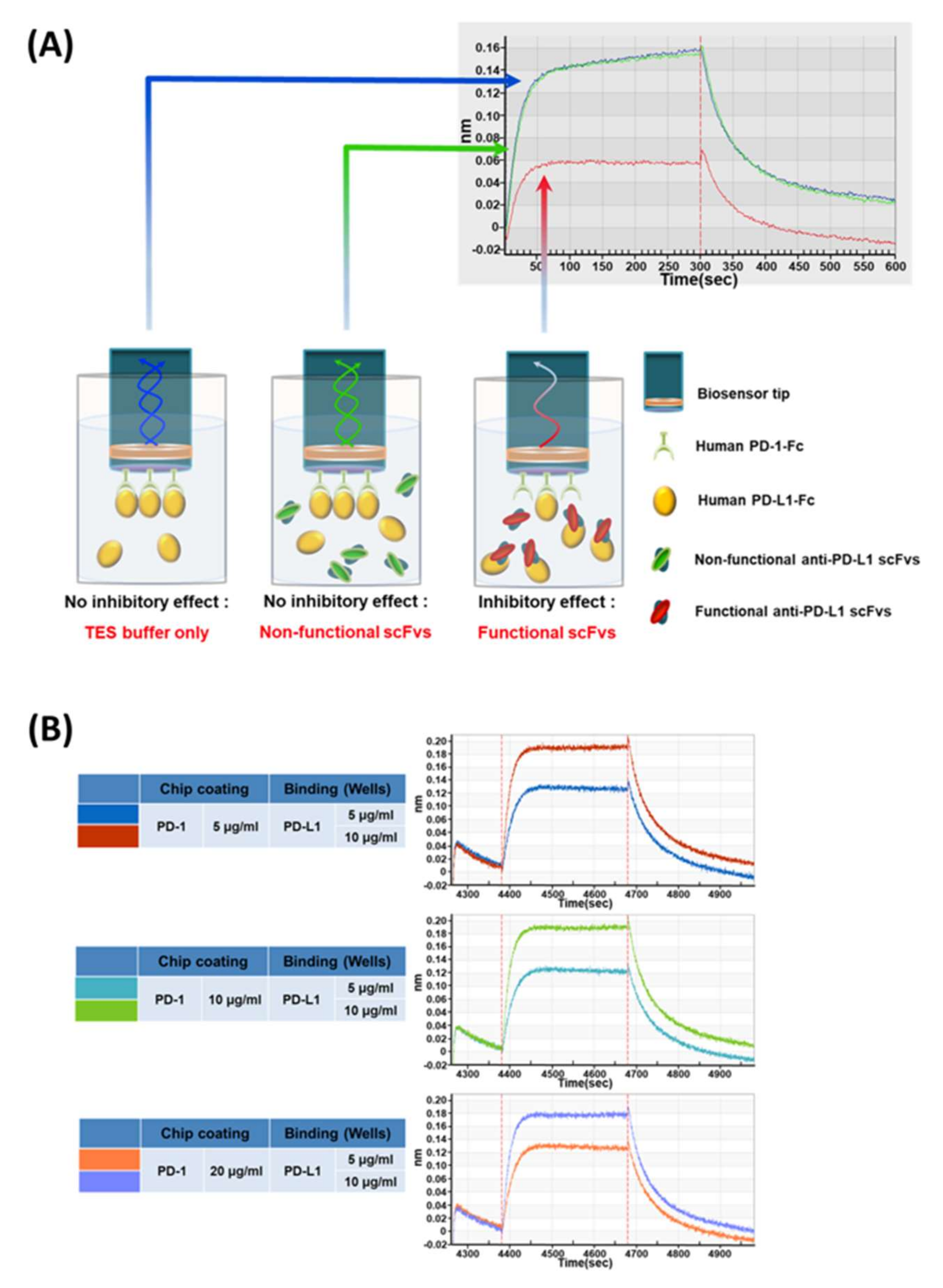
BLI-based PD-1/PD-L1 binding inhibition experiments were carried out by BLI using an Octet RED96 System (Pall ForteBio). The measurements were performed using amine-reactive second-generation (AR2G) biosensors. The human PD-1-Fc (Sino Biological Inc., Beijing, China) proteins were immobilized on the biosensor tip surface. The binding or dissociation of soluble human PD-L1-Fc proteins at the biosensor tip surface caused a pattern shift in the reflectance interference wave that is correlated with the changes in the mass or thickness of the biolayer at the sensor tip, which is denoted as a sensorgram. All steps were performed at 30 °C with shaking at 400 rpm in a black 96-well plate, with a working volume of 200 μL in each well. The human PD-1-Fc proteins were immobilized onto the AR2G biosensor surface via a 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide/N-hydroxysuccinimide (EDC/Sullfo-NHS)-mediated amine-coupling reaction, in accordance with the manufacturer’s directions. The human PD-1-Fc-immobilized AR2G biosensors were applied to the procedure of baseline (DW, 60 s), sensor activation (EDC/sulfo-NHS, 300 s), 5 µg/mL human PD-1-Fc protein loading (10 mM sodium acetate, pH 5, 1800 s), quenching (1 M ethanolamine, pH 8.5, 300 s), blocking (3% w/v BSA in PBS, 1800 s), and baseline (running buffer, 120 s). The human PD-L1-Fc protein (10 µg/mL, 100 μL) in the running buffer was mixed with 100 μL scFv-containing periplasmic crude extract, then applied for association for 300 s and dissociation (running buffer, 300 s). For the control sample, the same amount of TES buffer (0.5×, 100 μL) was added to the PD-L1 sample to remove the interference from the TES buffer itself. The response data were normalized using Octet data analysis software version 9.0.0.14 (Pall ForteBio).
2.9. PD-1/PD-L1 Blockade Bioassay
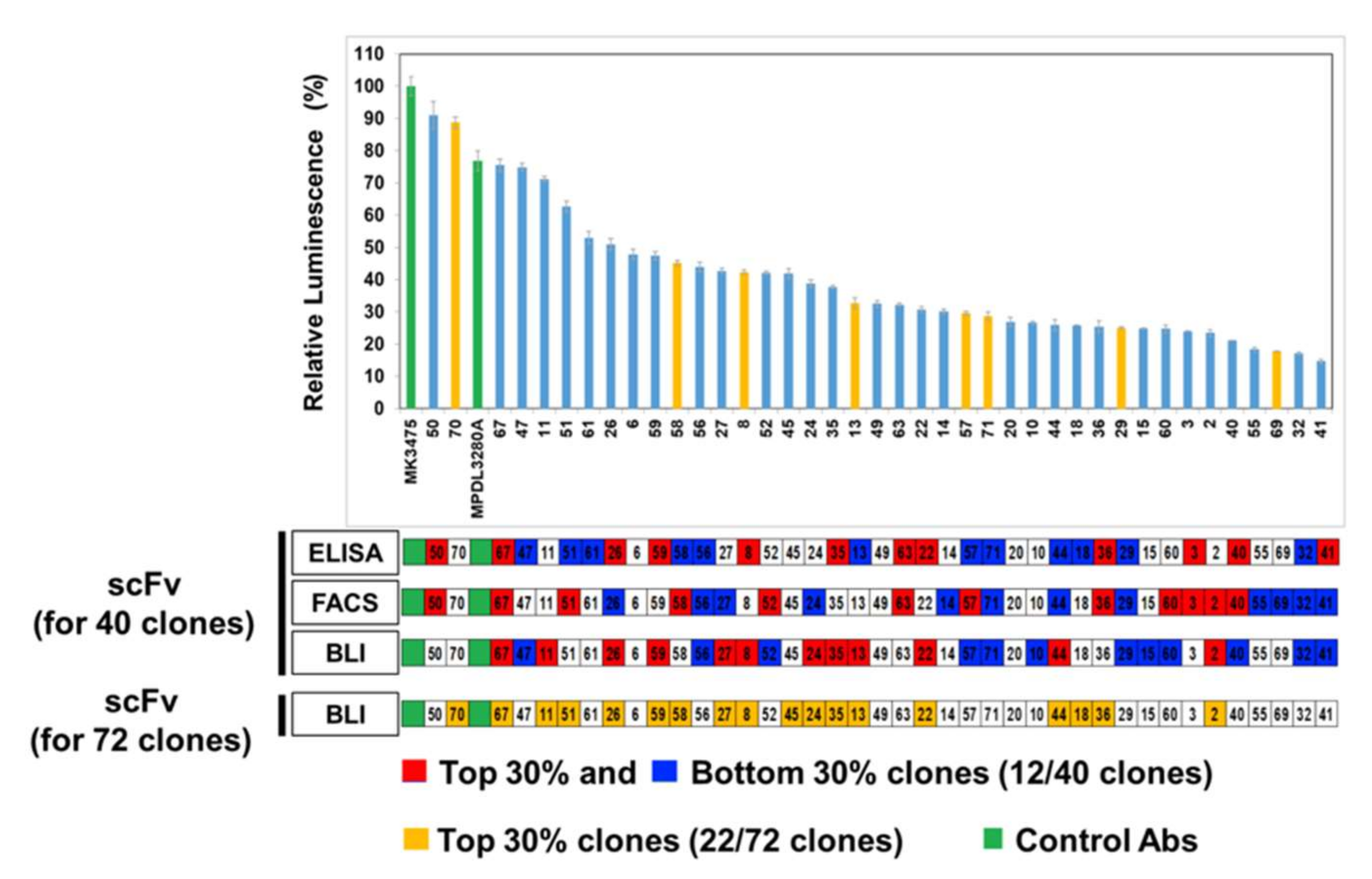
The inhibitory activity of the anti-PD-L1 antibody was analyzed by a luciferase reporter assay, performed in accordance with the manufacturer’s instructions (Promega, Fitchburg, WI, USA). PD-L1 aAPC/CHOK1 cells (4 × 105 cells/100 μL/well) in an F12 medium containing 10% FBS were seeded into white 96-well plates (Corning, Corning, NY, USA) and incubated for 17 h at 37 °C. After culturing the PD-L1 aAPC/CHOK1 cells, the medium was removed, and cells were pre-incubated with 10 µg/mL (40 μL/well) anti-PD-L1 antibodies or control antibodies (MK3475 or MPDL3280A) in RPMI1640 medium containing 2% FBS for 30 min at 37 °C. Then the PD-1 NFAT-luc/Jurkat T cells (5 × 105 cells/40 μL/well) in RPMI1640 medium containing 2% FBS were added to each well and co-cultured for 6 h at 37 °C. When co-cultured, inhibitor antibodies block the interaction of PD-1 and PD-L1, resulting in increasing NFAT-mediated luciferase activity in PD-1 NFAT-luc/Jurkat T cells. After the co-culturing, 80 μL Bio-Glo reagent was added and incubated for 7 min at room temperature (RT). The luminescence was measured using a microtiter plate reader (VICTOR X4, Perkin Elmer).
2.10. In Vivo MC38 Murine Tumor Models and Treatments
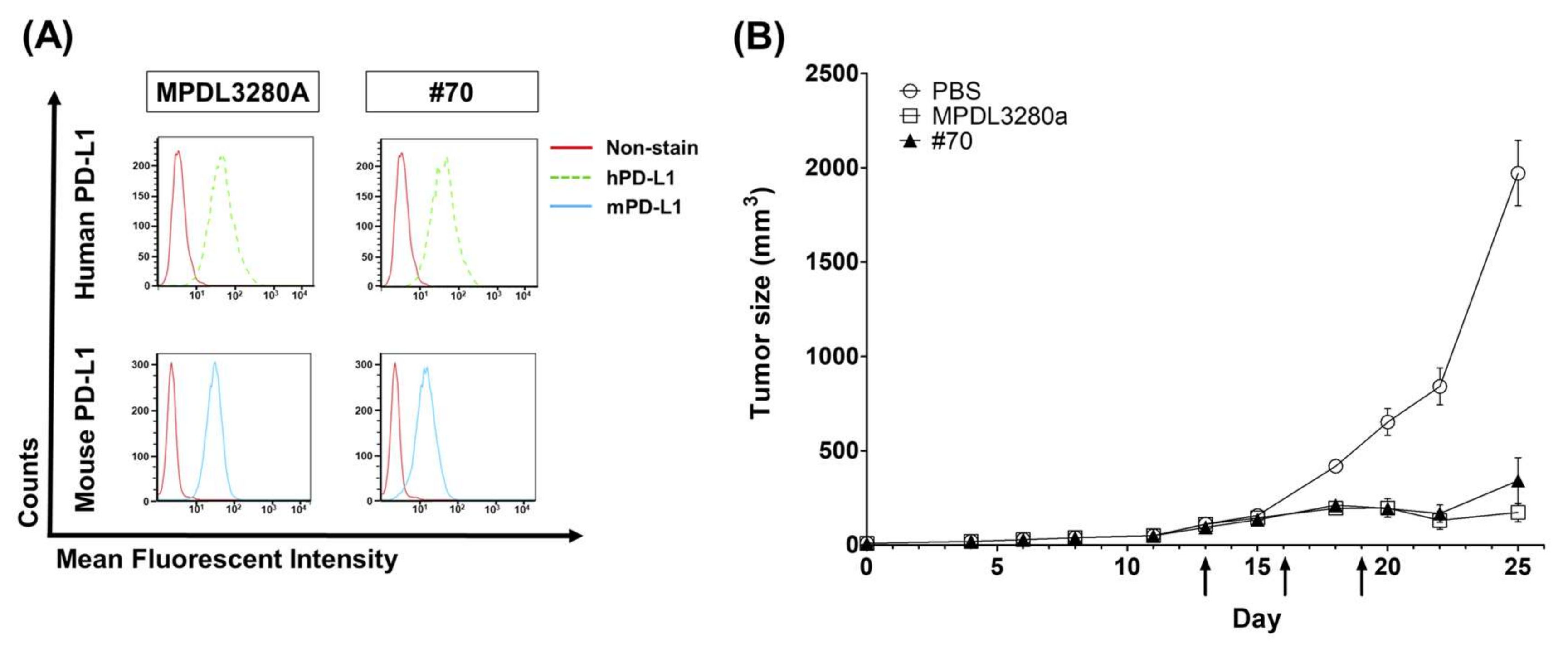
C57BL/6 mice were purchased from Orient Bio (Gyeonggi, Korea) and maintained under specific pathogen-free (SPF) conditions. All animal care and experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Kangwon National University (approval no. KW-171204-1). First, 5 × 105 MC38 tumor cells were subcutaneously (s.c.) injected into the flanks of C57BL/6 mice. When the average estimated tumor volume reached around 80 mm3 ± 20 (tumor volume = width2 × length × 0.5), the mice were randomly divided into five groups (n = 5 mice per group), and three doses of 200 µg (10 mg/kg) of anti-PD-L1 antibodies (MPDL3280 and #70) or PBS control were injected intraperitoneally (i.p.) every three days. Tumors were measured 2–3 times weekly. Mice were sacrificed when the tumor volume was ≥2000 mm3 and tumors were surgically isolated from tumor-bearing mice.
2.11. Statistical Analysis
Data collected from ELISA, flow cytometry, BLI, and the cell-based assay were plotted and analyzed using IBM SPSS statistics version 24.0 (IBM Corp., Armonk, NY, USA). For a correlation analysis between binding activity and inhibitory activity, we used the Pearson correlation analysis. The correlation between binding activity and inhibitory activity was confirmed by calculating the Pearson correlation coefficient. Data from in vivo experiments were analyzed with Prism (version 5.0, GraphPad software, La Jolla, CA, USA). Two-tailed Student’s t-tests were used to compare each of the antibody treatment groups with a single control. In analyses of in vivo data, the error bars represent standard deviation (SD).
4. Discussion
Therapeutic antibodies have become some of the most promising drugs for diverse diseases in the clinic. So far, 93 monoclonal antibodies have been FDA approved for therapy, and a number of candidates are in the advanced phases of clinical trials [
33,
34].
Phage display has been a fundamental, robust, and well-established technology for the development of therapeutic antibodies. In the process of phage display, the initial screening of antibodies has usually been performed by an ELISA assay using scFv, Fab, or the phage particle itself; a sequencing analysis follows to identify independent clones. Usually these independent clones are transformed into IgG-format antibodies for the functional characterization. When there are many independent clones, the transformation to the IgG format itself can be highly difficult work to most of the academic/industry researchers. Although the first ELISA assay can only show each antibody’s basic target binding, the following assays with IgG antibodies are related to the functional assessment of the clones, such as the verification of the target’s function change with other molecules, such as the receptor‒ligand interaction by antibody treatment. Therefore, if the assessment of the inhibitory function of the scFv antibody is possible in the earlier phase, prior to full IgG conversion, it would be highly beneficial to the whole process of developing therapeutic antibody candidates.
BLI-based assays have been very useful tools and the instrument has been a flexible high-throughput system for the antibody development process. It has been well used as a screening tool for high-level antibody-producing cell-line development [
35] and initial antibody selection [
36]. As an initial antibody screening tool for the hybridoma antibody selection, Lad et al. (2015) reported the strong benefit of the high-throughput kinetic screening of hybridoma antibodies, focusing on the dissociation rate (off-rate) using BLI technology [
36], which effectively screened out high-affinity antibodies that could be ignored by typical ELISA-based hybridoma antibody screening. However, this screening method still cannot verify the functionality of isolated antibodies from the screening unless there are additional functional assays.
In our study, we developed new BLI-based PD-1/PD-L1 inhibition assays that can be applicable to periplasmic scFv antibodies, in which the direct interaction between target (PD-L1) and receptor (PD-1) can be measured first; then any sensorgram changes from the addition of the antibody candidate to the target and receptor interaction are compared. Our study focused more on the assessment of the functionality of antibody clones from the phage-display antibody screening, which indicated the inhibition of the target protein’s (in our case, PD-L1) interaction with its partner (PD-1). With the help of an innovative instrument, the Octet RED96 system from ForteBio, we were able to set up the experimental conditions using a 96-well-type inhibition assay for the PD-1‒PD-L1 interaction. When we applied the assay, even to the unnormalized periplasmic scFv antibodies, PD-L1 incubated with antibodies clearly showed the reduced sensorgrams that represented the inhibitory functions of antibody clones, while the negative control antibody did not show such a sensorgram change.
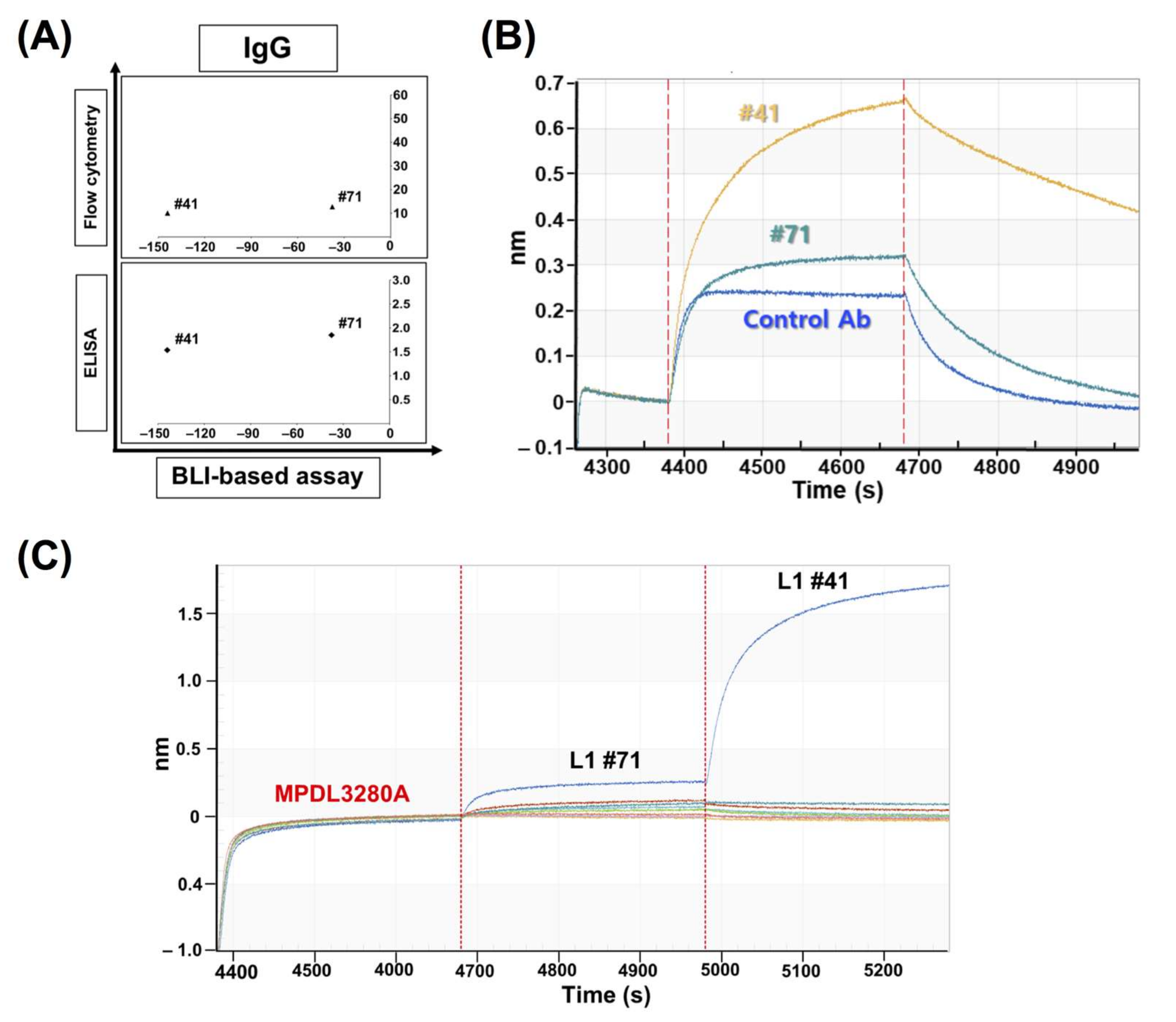
In comparison with the results from the cell-based inhibition assay with IgG antibodies, the scFv clones with high inhibition (the top 30% group) were mostly included in the set of highly functional antibodies in the cell-based assay, in contrast to the high scFv binders from the ELISA or FACS assay, which means that the BLI-based functional assay can be very informative in candidate selection and increase the probability of functional antibody development at the initial screening stage. When the correlation between the cell-based assay and ELISA/FACS/BLI-based assay with scFv antibodies was analyzed, there was no statistically meaningful correlation, which could be because of the unnormalized scFvs, which is usually the main problem with primary antibody screening. It is difficult to make scFv expression even in the screening when there are so many clones in ELISA and other assays. However, IgG-format antibodies showed a relatively high level of correlation between the cell-based assay and the ELISA/FACS/BLI assay. In particular, the FACS and BLI-based assays showed strong correlations with the cell-based assay (r = 0.931, p = 3.13 × 10−18, and r = 0.772, p = 1.14 × 10−8), which confirmed the utility of BLI-based inhibition assays in functional antibody development.
As for the higher correlation of the FACS assay with the in vitro cell-based assay in our IgG case, in theory, the high FACS shift represents the high-affinity antibody binding to the overexpressed antigens or well-exposed epitopes in a native conformation, but such a high FACS shift does not always prove the functionality of the epitopes. So, it is a very interesting result from our experiment, but it cannot be generalized so as to claim that the FACS assay has in most cases a strong correlation with the in vitro cell-based functional assay. However, for the BLI assay, the sensorgram shows the relative bindings between target protein (PD-L1) and counterpart (PD-1) with or without the inhibitory antibodies, which in turn represent the functionality of the antibodies on the target’s interaction. So, even though its correlation is slightly weaker than in the FACS assay in the current experiment, the fact that the BLI assay has a correlation with the in vitro functional assay itself is an important validation of the feasibility of using the BLI-based assay as a functional assay tool in therapeutic antibody development. As to the higher correlation of the FACS assay with an in vitro assay in our results, we reasoned that our phage-display panning against PD-L1 had generated, in some way, many antibodies targeting the epitopes that overlapped with the PD-1 binding site and so was very sensitive on their conformation (the ELISA results did not show such correlations).
When we looked at the covariation between the ELISA, FACS, and BLI-based inhibition assays, there were no statistically meaningful covariations, which means they showed different features of scFv antibodies at the first screening step after phage display and the BLI-based assay was important to evaluate the functionality of the antibody clones. Interestingly, a lot of low-inhibition scFv clones showed a low level of expression in mammalian systems, so most of them could not be tested for a cell-based assay. This could be because the low inhibition activities of scFv come from nonfunctional properties or the low expression of individual clones; however, we cannot conclude which factor contributed most.
For testing in vivo antibody activity, especially for the PD-L1-targeting antibody, it was difficult to compare the BLI assay to in vivo functionality because only human‒mouse cross-species reactive antibodies are eligible for an in vivo assay. Human‒mouse cross-reactive clone #70 was chosen as an in vivo test clone. Moderate inhibition activity in a BLI assay using scFv was selected for the in vivo animal model study and showed strong anticancer inhibition activity, together with Genentech’s MPDL3280A control antibody. To become a final therapeutic antibody candidate, further analysis of various parameters such as the stability, expression, and solubility of the antibody will be needed.
Clones #41 and #71 showed very interesting results: BLI-based sensorgrams showed increased peaks compared to the control sensorgram. Later, through the competition ELISA and BLI assays, they showed compatible binding to PD-L1, together with MPDL3280A, which means their epitopes are relatively far from the control antibody’s binding site. So, the clones showing these increased sensorgrams can be distinguished from the other clones and this categorization of two antibody groups with regard to their binding compatibility with the native ligand (PD-1) can be useful to the screening of at least two groups of antibodies for a sandwich-type antigen capture system or a multiple epitope targeting system.
The BLI-based functional assay has several advantages in phage-display screening: (1) It can be easily applied to a high-throughput screening system, so an automated functional assay at primary antibody screening in phage display can generate a highly efficient antibody screening process; (2) the qualitative assessment of functional antibody development from phage panning can be analyzed even at the initial scFv screening stage, and a BLI-based assay with an IgG antibody will provide highly reliable data for the development of therapeutic antibodies; and (3) it will be a highly efficient method to develop diagnostic antibody pairs for companion diagnostics.
Overall, we designed a new BLI-based functional assay for initial scFv screening and tried it out to screen functional antibodies’ PD-L1 targeting. A BLI-based assay in earlier stages showed a higher functional correlation with a later in vitro cell-based functional assay. That result correlated well with the results of an in vivo syngeneic mouse model study. Our study showed that the BLI-based assay is highly beneficial for functional antibody screening at earlier steps and for drug candidate development.

 and
and 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}