Discovery of Two Novel Negeviruses in a Dungfly Collected from the Arctic


 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation and RNA Extraction
2.2. Transcriptome and sRNA Sequencing
2.3. Host Insect Identification
2.4. Virus Discovery and Confirmation by Reverse Transcription-PCR (RT-PCR)
2.5. Determination of Viral Genome Termini and Transcript Abundance
2.6. Genome Annotation
2.7. Small RNA Analysis
2.8. Prevalence of Nege-Like Viruses Were Investigated in Invertebrates
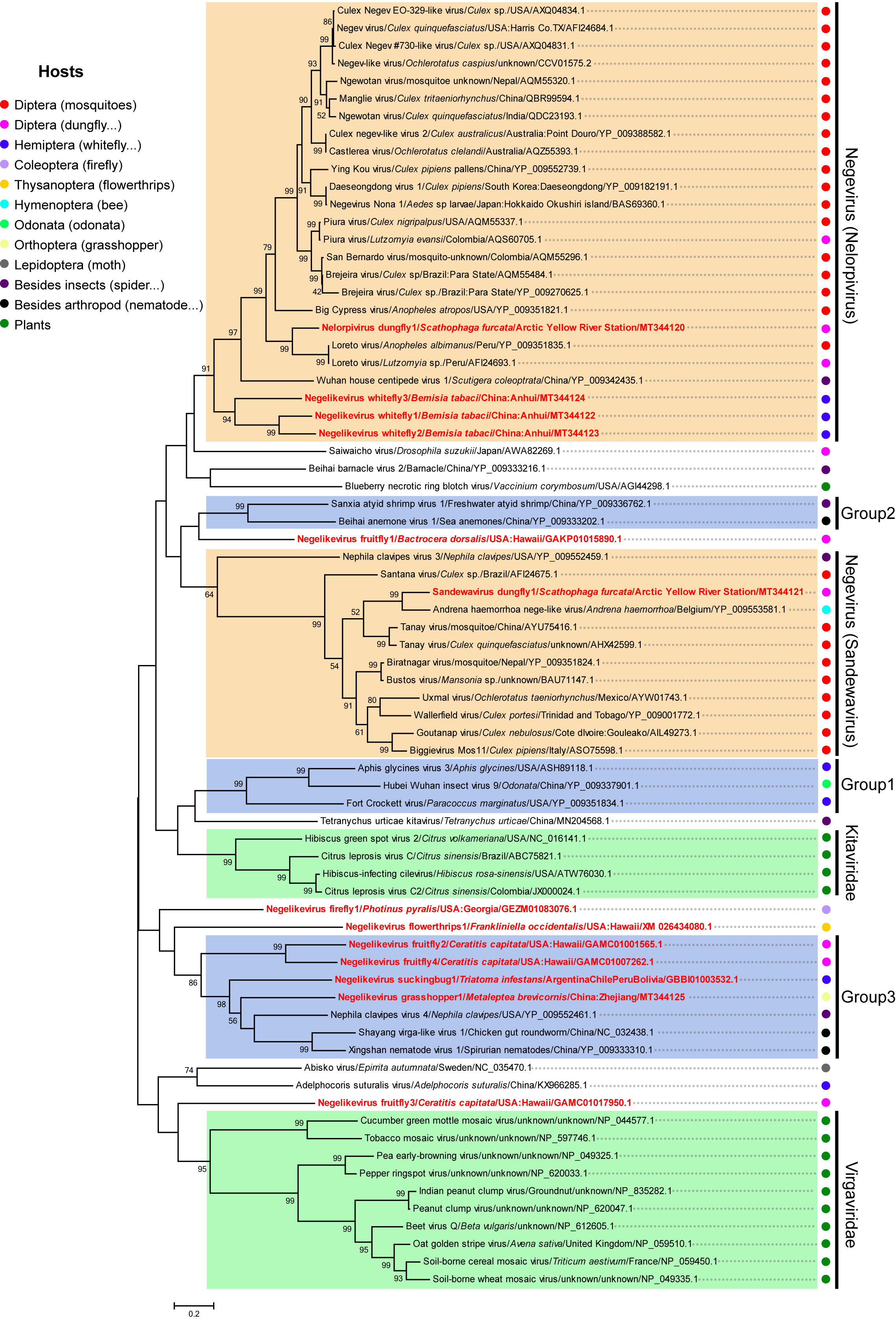
2.9. Phylogenetic Analyses
3. Results
3.1. Negeviruses Identified in Dungfly
3.2. Genome Organization of NVD1 and SVD1
3.3. NVD1 and SVD1 Are Targeted by the Host siRNA-Based Antiviral RNAi Pathway
3.4. The Presence of Further Nege-Like Virus Sequences in Public Databases Suggests That They Occur in Many Different Insects
3.5. Putative New Phylogenetic Clades and Host Diversity of Negeviruses in Invertebrates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vasilakis, N.; Tesh, R.B. Insect-specific viruses and their potential impact on arbovirus transmission. Curr. Opin. Virol. 2015, 15, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junglen, S.; Drosten, C. Virus discovery and recent insights into virus diversity in arthropods. Curr. Opin. Microbiol. 2013, 16, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Bolling, B.G.; Weaver, S.C.; Tesh, R.B.; Vasilakis, N. Insect-Specific Virus Discovery: Significance for the Arbovirus Community. Viruses 2015, 7, 4911–4928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marklewitz, M.; Zirkel, F.; Kurth, A.; Drosten, C.; Junglen, S. Evolutionary and phenotypic analysis of live virus isolates suggests arthropod origin of a pathogenic RNA virus family. Proc. Natl. Acad. Sci. USA 2015, 112, 7536–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasilakis, N.; Forrester, N.L.; Palacios, G.; Nasar, F.; Savji, N.; Rossi, S.L.; Guzman, H.; Wood, T.G.; Popov, V.; Gorchakov, R.; et al. Negevirus: A proposed new taxon of insect-specific viruses with wide geographic distribution. J. Virol. 2013, 87, 2475–2488. [Google Scholar] [CrossRef] [Green Version]
- Kallies, R.; Kopp, A.; Zirkel, F.; Estrada, A.; Gillespie, T.R.; Drosten, C.; Junglen, S. Genetic characterization of goutanap virus, a novel virus related to negeviruses, cileviruses and higreviruses. Viruses 2014, 6, 4346–4357. [Google Scholar] [CrossRef]
- Kawakami, K.; Kurnia, Y.W.; Fujita, R.; Ito, T.; Isawa, H.; Asano, S.; Binh, N.D.; Bando, H. Characterization of a novel negevirus isolated from Aedes larvae collected in a subarctic region of Japan. Arch. Virol. 2016, 161, 801–809. [Google Scholar] [CrossRef]
- Kuchibhatla, D.B.; Sherman, W.A.; Chung, B.Y.; Cook, S.; Schneider, G.; Eisenhaber, B.; Karlin, D.G. Powerful sequence similarity search methods and in-depth manual analyses can identify remote homologs in many apparently “orphan” viral proteins. J. Virol. 2014, 88, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Carapeta, S.; do Bem, B.; McGuinness, J.; Esteves, A.; Abecasis, A.; Lopes, A.; de Matos, A.P.; Piedade, J.; de Almeida, A.P.; Parreira, R. Negeviruses found in multiple species of mosquitoes from southern Portugal: Isolation, genetic diversity, and replication in insect cell culture. Virology 2015, 483, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Gu, Q.; Niu, J.; Wang, J.J. The RNA Virome and Its Dynamics in an Invasive Fruit Fly, Bactrocera dorsalis, Imply Interactions Between Host and Viruses. Microb. Ecol. 2020, 1–12. [Google Scholar] [CrossRef]
- Nabeshima, T.; Inoue, S.; Okamoto, K.; Posadas-Herrera, G.; Yu, F.; Uchida, L.; Ichinose, A.; Sakaguchi, M.; Sunahara, T.; Buerano, C.C.; et al. Tanay virus, a new species of virus isolated from mosquitoes in the Philippines. J. Gen. Virol. 2014, 95 Pt 6, 1390–1395. [Google Scholar] [CrossRef]
- Wang, J.; Wu, J.; Li, N.; Cao, Y.; He, Y.; Lin, J.; Li, H. A new Tanay virus isolated from mosquitoes in Guangxi, China. Arch. Virol. 2018, 163, 3177–3180. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; McLean, B.J.; Colmant, A.M.G.; Harrison, J.J.; Hall-Mendelin, S.; van den Hurk, A.F.; Johansen, C.A.; Watterson, D.; Bielefeldt-Ohmann, H.; Newton, N.D.; et al. Discovery and Characterisation of Castlerea Virus, a New Species of Negevirus Isolated in Australia. Evol. Bioinform. 2017, 13, 1176934317691269. [Google Scholar]
- Auguste, A.J.; Carrington, C.V.; Forrester, N.L.; Popov, V.L.; Guzman, H.; Widen, S.G.; Wood, T.G.; Weaver, S.C.; Tesh, R.B. Characterization of a novel Negevirus and a novel Bunyavirus isolated from Culex (Culex) declarator mosquitoes in Trinidad. J. Gen. Virol. 2014, 95 Pt 2, 481–485. [Google Scholar] [CrossRef] [Green Version]
- Nunes, M.R.T.; Contreras-Gutierrez, M.A.; Guzman, H.; Martins, L.C.; Barbirato, M.F.; Savit, C.; Balta, V.; Uribe, S.; Vivero, R.; Suaza, J.D.; et al. Genetic characterization, molecular epidemiology, and phylogenetic relationships of insect-specific viruses in the taxon Negevirus. Virology 2017, 504, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Mwaliko, C.; Atoni, E.; Wang, Y.; Zhang, Y.; Zhan, J.; Hu, X.; Xia, H.; Yuan, Z. Characterization of a Novel Tanay Virus Isolated From Anopheles sinensis Mosquitoes in Yunnan, China. Front. Microbiol. 2019, 10, 1963. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Gonzalez, P.L.; Dos Santos, G.F.; Chabi-Jesus, C.; Harakava, R.; Kitajima, E.W.; Freitas-Astua, J. Passion Fruit Green Spot Virus Genome Harbors a New Orphan ORF and Highlights the Flexibility of the 5′-End of the RNA2 Segment Across Cileviruses. Front. Microbiol. 2020, 11, 206. [Google Scholar] [CrossRef]
- Schoonvaere, K.; Smagghe, G.; Francis, F.; de Graaf, D.C. Study of the Metatranscriptome of Eight Social and Solitary Wild Bee Species Reveals Novel Viruses and Bee Parasites. Front. Microbiol. 2018, 9, 177. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. elife 2015, 4, e05378. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Andika, I.B.; Shen, J.; Lv, Y.; Ji, Y.; Sun, L.; Chen, J. Characterization of rice black-streaked dwarf virus- and rice stripe virus-derived siRNAs in singly and doubly infected insect vector Laodelphax Striatellus. PLoS ONE 2013, 8, e66007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Ding, S.W.; Voinnet, O. Antiviral immunity directed by small RNAs. Cell 2007, 130, 413–426. [Google Scholar] [CrossRef] [Green Version]
- Mi, S.; Cai, T.; Hu, Y.; Chen, Y.; Hodges, E.; Ni, F.; Wu, L.; Li, S.; Zhou, H.; Long, C. Sorting of Small RNAs into Arabidopsis Argonaute Complexes Is Directed by the 5′ Terminal Nucleotide. Cell 2008, 133, 116–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debat, H.J. An RNA Virome Associated to the Golden Orb-Weaver Spider Nephila clavipes. Front. Microbiol. 2017, 8, 2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Z.X.; Huang, H.J.; Lu, G.; Zhuo, J.C.; Chen, J.P.; Zhang, C.X.; Li, J.M. Virome analysis of whitefly collected from Anhui, China. Manuscript in preparation.
- Ohlund, P.; Hayer, J.; Lunden, H.; Hesson, J.C.; Blomstrom, A.L. Viromics Reveal a Number of Novel RNA Viruses in Swedish Mosquitoes. Viruses 2019, 11, 1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, C.L.; Longdon, B.; Lewis, S.H.; Obbard, D.J. Twenty-Five New Viruses Associated with the Drosophilidae (Diptera). Evol. Bioinform. 2016, 12 (Suppl. 2), 13–25. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Neville, P.; Nicholson, J.; Eden, J.S.; Imrie, A.; Holmes, E.C. High-Resolution Metatranscriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Geib, S.M.; Calla, B.; Hall, B.; Hou, S.; Manoukis, N.C. Characterizing the developmental transcriptome of the oriental fruit fly, Bactrocera dorsalis (Diptera: Tephritidae) through comparative genomic analysis with Drosophila melanogaster utilizing modENCODE datasets. BMC Genom. 2014, 15, 942. [Google Scholar] [CrossRef] [Green Version]
- Calla, B.; Hall, B.; Hou, S.; Geib, S.M. A genomic perspective to assessing quality of mass-reared SIT flies used in Mediterranean fruit fly (Ceratitis capitata) eradication in California. BMC Genom. 2014, 15, 98. [Google Scholar] [CrossRef] [Green Version]
- Medd, N.C.; Fellous, S.; Waldron, F.M.; Xuereb, A.; Nakai, M.; Cross, J.V.; Obbard, D.J. The virome of Drosophila suzukii, an invasive pest of soft fruit. Virus Evol. 2018, 4, vey009. [Google Scholar] [CrossRef]
- Schwarz, A.; Medrano-Mercado, N.; Schaub, G.A.; Struchiner, C.J.; Bargues, M.D.; Levy, M.Z.; Ribeiro, J.M. An updated insight into the Sialotranscriptome of Triatoma infestans: Developmental stage and geographic variations. PLoS Negl. Trop. Dis. 2014, 8, e3372. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Krueger, E.N.; Liu, S.; Dorman, K.; Bonning, B.C.; Miller, W.A. Discovery of known and novel viral genomes in soybean aphid by deep sequencing. Phytobiomes 2017, 1, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xu, P.; Yang, X.; Yuan, H.; Chen, L.; Lu, Y. The genome sequence of a novel RNA virus in Adelphocoris suturalis. Arch. Virol. 2017, 162, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- Al-Wathiqui, N.; Fallon, T.R.; South, A.; Weng, J.K.; Lewis, S.M. Molecular characterization of firefly nuptial gifts: A multi-omics approach sheds light on postcopulatory sexual selection. Sci. Rep. 2016, 6, 38556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Miranda, J.R.; Hedman, H.; Onorati, P.; Stephan, J.; Karlberg, O.; Bylund, H.; Terenius, O. Characterization of a novel RNA virus discovered in the autumnal moth Epirrita autumnata in Sweden. Viruses 2017, 9, 214. [Google Scholar] [CrossRef]
- Roundy, C.M.; Azar, S.R.; Rossi, S.L.; Weaver, S.C.; Vasilakis, N. Insect-Specific Viruses: A Historical Overview and Recent Developments. Adv. Virus Res. 2017, 98, 119–146. [Google Scholar] [PubMed]
- Kondo, H.; Chiba, S.; Maruyama, K.; Andika, I.B.; Suzuki, N. A novel insect-infecting virga/nege-like virus group and its pervasive endogenization into insect genomes. Virus Res. 2019, 262, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Lenz, O.; Pribylova, J.; Franova, J.; Koloniuk, I. Fragaria vesca-associated virus 1, a new virus related to negeviruses. Arch. Virol. 2020, 165, 1249–1252. [Google Scholar] [CrossRef]
- Moreira, L.A.; Iturbe-Ormaetxe, I.; Jeffery, J.A.; Lu, G.; Pyke, A.T.; Hedges, L.M.; Rocha, B.C.; Hall-Mendelin, S.; Day, A.; Riegler, M.; et al. A Wolbachia symbiont in Aedes aegypti limits infection with dengue, Chikungunya, and Plasmodium. Cell 2009, 139, 1268–1278. [Google Scholar] [CrossRef] [Green Version]
- Rances, E.; Ye, Y.H.; Woolfit, M.; McGraw, E.A.; O’Neill, S.L. The relative importance of innate immune priming in Wolbachia-mediated dengue interference. PLoS Pathog 2012, 8, e1002548. [Google Scholar] [CrossRef] [Green Version]
- Hobson-Peters, J.; Yam, A.W.Y.; Lu, J.W.F.; Setoh, Y.X.; May, F.J.; Kurucz, N.; Walsh, S.; Prow, N.A.; Davis, S.S.; Weir, R. A new insect-specific flavivirus from northern Australia suppresses replication of West Nile virus and Murray Valley encephalitis virus in co-infected mosquito cells. PLoS ONE 2013, 8, e56534. [Google Scholar] [CrossRef]
- Kenney, J.L.; Solberg, O.D.; Langevin, S.A.; Brault, A.C. Characterization of a novel insect-specific flavivirus from Brazil: Potential for inhibition of infection of arthropod cells with medically important flaviviruses. J. Gen. Virol. 2014, 95 Pt 12, 2796–2808. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Tentative Name | NCBI Accession No. | Length | E-Value | Negevirus Protein | Query Start | Query End | Homology Virus | RdRP # Identity | Homology Virus Reference |
|---|---|---|---|---|---|---|---|---|---|
| Negelikevirus firefly1 | GEZM01083076.1 | 5125 | 6 × 10−154 | ORF1 (partial) | 4361 | 1611 | Hammarskog virga-like virus | 40% | [34] |
| Negelikevirus flowerthrips1 | XM_026434080.1 | 1791 | 7 × 10−76 | ORF1 (partial) | 247 | 1734 | Buckhurst virus | 38% | [35] |
| Negelikevirus suckingbug1 | GBBI01003532.1 | 1488 | 8 × 10−119 | ORF1 (partial) | 10 | 1485 | Hubei virga-like virus 16 | 46% | [19] |
| Negelikevirus fruitfly1 | GAKP01015890.1 | 6984 | 0 | ORF1 (Complete) | 44 | 4492 | Hammarskog virga-like virus | 51% | [34] |
| Negelikevirus fruitfly2 | GAMC01001565.1 | 10,007 | 0 | ORF1 (Complete) | 71 | 6526 | Aedes camptorhynchus negev-like virus | 62% | [36] |
| Negelikevirus fruitfly3 | GAMC01017950.1 | 9925 | 0 | ORF1 (Complete) | 112 | 9870 | Blackford virus | 61% | [35] |
| Negelikevirus fruitfly4 | GAMC01007262.1 | 10,264 | 0 | ORF1 (Complete) | 64 | 6531 | Aedes camptorhynchus negev-like virus | 54% | [36] |
| Virus Name/Tentative Name | Host Information | Location/Year | GenBank Accession No. | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|
| Hosts/Species | Phylum | Class | Order | Family | Genus | ||||
| Nelorpivirus dungfly1 | Scathophaga furcata | Arthropoda | Insecta | Diptera | Scathophagidae | Scathophaga | Arctic yellow river station/2019 | MT344120 | This study |
| Sandewavirus dungfly1 | MT344121 | ||||||||
| Negelikevirus fruitfly1 | Bactrocera dorsalis | Arthropoda | Insecta | Diptera | Tryetidae | Bactrocera | Puna, Hawaii, USA/1984 | GAKP01015890.1 | [37] |
| Negelikevirus fruitfly2 | Ceratitis capitata | Arthropoda | Insecta | Diptera | Tephritidae | Ceratitis | Waimanalo, Hawaii, USA/NA | GAMC01001565.1 | [38] |
| Negelikevirus fruitfly3 | GAMC01017950.1 | ||||||||
| Negelikevirus fruitfly4 | GAMC01007262.1 | ||||||||
| Piura virus | Lutzomyia evansi | Arthropoda | Insecta | Diptera | Psychodidae | Lutzomyia | Colombia/2013 | AQS60705.1 | [15] |
| Loreto virus | Lutzomyia sp. | Arthropoda | Insecta | Diptera | Psychodidae | Lutzomyia | Peru/1977 | AFI24693.1 | [5] |
| Saiwaicho virus | Drosophila suzukii | Arthropoda | Insecta | Diptera | Drosophilidae | Drosophila | Japan/2016 | AWA82269.1 | [39] |
| Negelikevirus whitefly1 | Bemisia tabaci | Arthropoda | Insecta | Hemiptera | Aleyrodidae | Bemisia | Anhui, China/2019 | MT344122 | [33] |
| Negelikevirus whitefly2 | MT344123 | ||||||||
| Negelikevirus whitefly3 | MT344124 | ||||||||
| Negelikevirus suckingbug1 | Triatoma infestans | Arthropoda | Insecta | Hemiptera | Reduviidae | Triatoma | Argentina/2007, Chile/1979, Peru/2008, Bolivia/2003–2012 | GBBI01003532.1 | [40] |
| Fort Crockett virus | Paracoccus marginatus | Arthropoda | Insecta | Hemiptera | Pseudococcidae | Paracoccus | USA/2015 | YP_009351834.1 | [15] |
| Aphis glycines virus 3 | Aphis glycines | Arthropoda | Insecta | Hemiptera | Aphididae | Aphis | USA/2009 | ASH89118.1 | [41] |
| Adelphocoris suturalis virus | Adelphocoris suturalis | Arthropoda | Insecta | Hemiptera | Miridae | Adelphocoris | China/2015 | KX966285.1 | [42] |
| Negelikevirus firefly1 | Photinus pyralis | Arthropoda | Insecta | Coleoptera | Lampyridae | Photinus | Lawrenceville, Georgia, USA/2015 | GEZM01083076.1 | [43] |
| Negelikevirus flowerthrips1 | Frankliniella occidentalis | Arthropoda | Insecta | Thysanoptera | Thripidae | Frankliniella | Oahu, Hawaii, USA/NA | XM_026434080.1 | NA |
| Andrena haemorrhoa nege-like virus | Andrena haemorrhoa | Arthropoda | Insecta | Hymenoptera | Andrenidae | Andrena | Belgium/2015 | YP_009553581.1 | [18] |
| Hubei Wuhan insect virus 9 | Odonata | Arthropoda | Insecta | Odonata | - | - | China/2013 | YP_009337901.1 | [19] |
| Negelikevirus grasshopper1 | Metaleptea brevicornis | Arthropoda | Insecta | Orthoptera | Acrididae | Metaleptea | Huzhou, Zhejiang, China/2019 | MT344125 | [33] |
| Abisko virus | Epirrita autumnata | Arthropoda | Insecta | Lepidoptera | Geometridae | Epirrita | Sweden/2013 | NC_035470.1 | [44] |
| Nephila clavipes virus 3 | Nephila clavipes | Arthropoda | Arachnida | Araneae | Nephilidae | Nephila | USA/2013 | YP_009552459.1 | [32] |
| Nephila clavipes virus 4 | Nephila clavipes | Arthropoda | Arachnida | Araneae | Nephilidae | Nephila | USA/2013 | YP_009552461.1 | [32] |
| Tetranychus urticae kitavirus | Tetranychus urticae | Arthropoda | Arachnida | Trombidiformes | Tetranychidae | Tetranychus | China/2018 | MN204568.1 | NA |
| Sanxia atyid shrimp virus 1 | Freshwater atyid shrimp | Arthropoda | Malacostraca | Decapoda | Atyidae | - | China/2014 | YP_009336762.1 | [19] |
| Beihai barnacle virus 2 | Barnacle | Arthropoda | Maxillopoda | - | - | - | China/2014 | YP_009333216.1 | [19] |
| Wuhan house centipede virus 1 | Scutigera coleoptrata | Arthropoda | Chilopoda | Scutigeromorpha | Scutigeridae | Scutigera | China/2013 | YP_009342435.1 | [19] |
| Shayang virga-like virus 1 | Chicken gut roundworm | Nematode | Secernentea | Ascaridida | Ascaridiidae | - | China/2014 | NC_032438.1 | [19] |
| Xingshan nematode virus 1 | Spirurian nematodes | Nematode | Chromadorea | Rhabditida | - | - | China/2014 | YP_009333310.1 | [19] |
| Beihai anemone virus 1 | Sea anemones | Cnidaria | Anthozoa | Actiniaria | - | - | China/2014 | YP_009333202.1 | [19] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, G.; Ye, Z.-X.; He, Y.-J.; Zhang, Y.; Wang, X.; Huang, H.-J.; Zhuo, J.-C.; Sun, Z.-T.; Yan, F.; Chen, J.-P.; et al. Discovery of Two Novel Negeviruses in a Dungfly Collected from the Arctic. Viruses 2020, 12, 692. https://doi.org/10.3390/v12070692
Lu G, Ye Z-X, He Y-J, Zhang Y, Wang X, Huang H-J, Zhuo J-C, Sun Z-T, Yan F, Chen J-P, et al. Discovery of Two Novel Negeviruses in a Dungfly Collected from the Arctic. Viruses. 2020; 12(7):692. https://doi.org/10.3390/v12070692
Chicago/Turabian StyleLu, Gang, Zhuang-Xin Ye, Yu-Juan He, Yan Zhang, Xin Wang, Hai-Jian Huang, Ji-Chong Zhuo, Zong-Tao Sun, Fei Yan, Jian-Ping Chen, and et al. 2020. "Discovery of Two Novel Negeviruses in a Dungfly Collected from the Arctic" Viruses 12, no. 7: 692. https://doi.org/10.3390/v12070692
APA StyleLu, G., Ye, Z. -X., He, Y. -J., Zhang, Y., Wang, X., Huang, H. -J., Zhuo, J. -C., Sun, Z. -T., Yan, F., Chen, J. -P., Zhang, C. -X., & Li, J. -M. (2020). Discovery of Two Novel Negeviruses in a Dungfly Collected from the Arctic. Viruses, 12(7), 692. https://doi.org/10.3390/v12070692