Strain-Specific Epigenetic Regulation of Endogenous Retroviruses: The Role of Trans-Acting Modifiers

Abstract
:1. Introduction
2. Epigenetic Regulation of ERVs
3. Evidence of Strain-Specific ERV Control
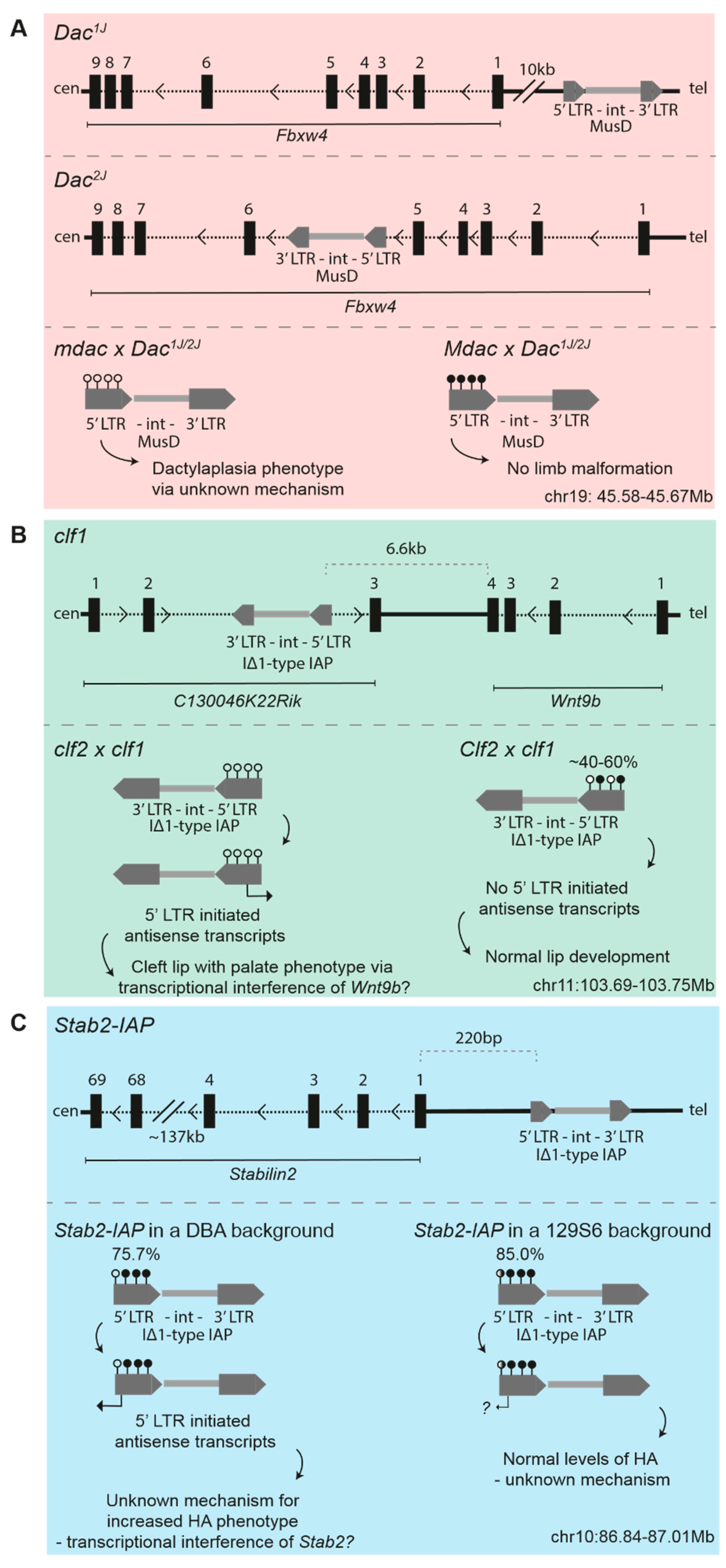
3.1. Dactylaplasia-Causing MusD Insertions at Fbxw4
3.2. Cleft Lip Palate-Causing IAP Insertions at Wnt9b
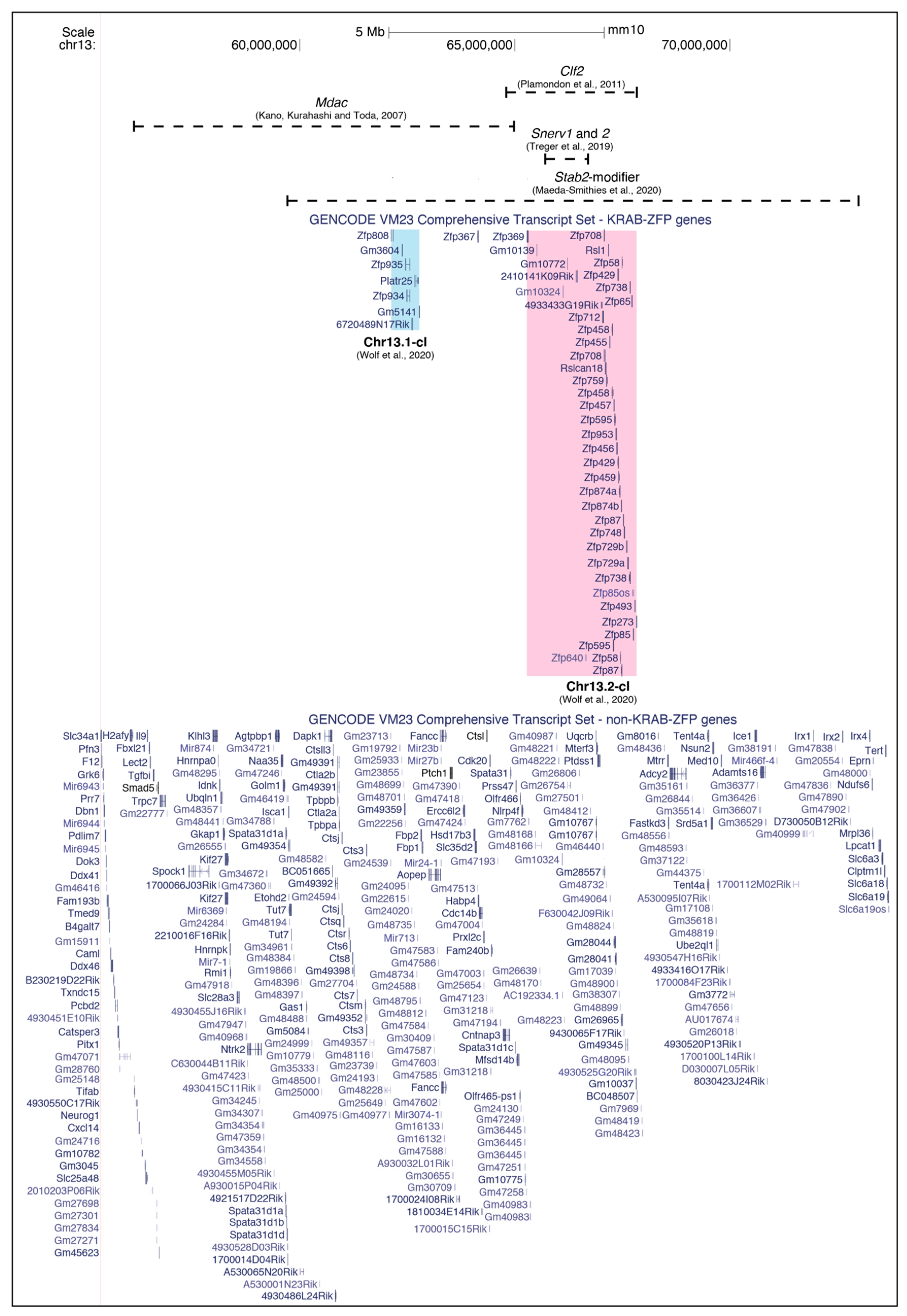
3.3. Non-Ecotropic ERV Activation Links to Lupus
3.4. IAP-Driven Stabilin2 Expression in DBA/2J Mice
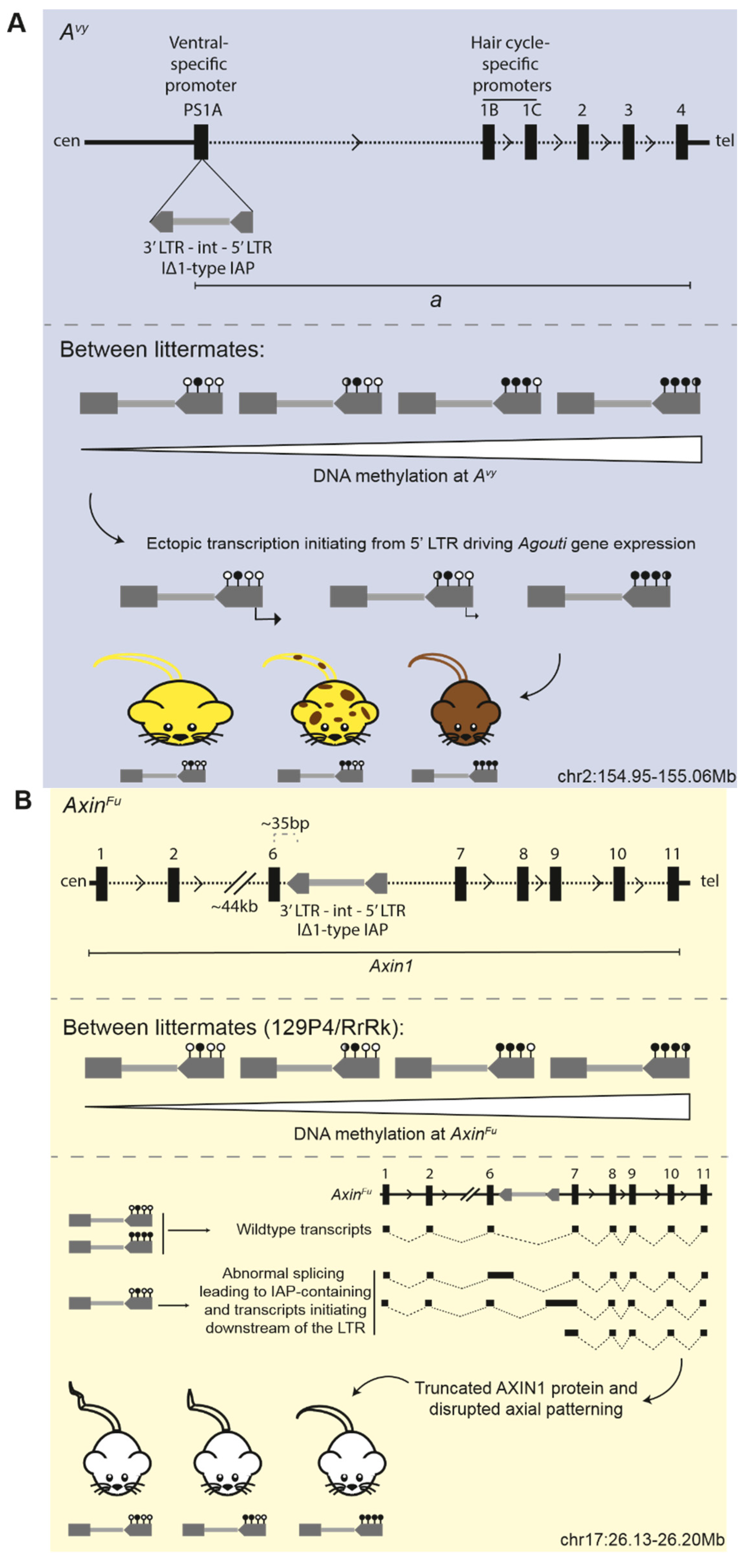
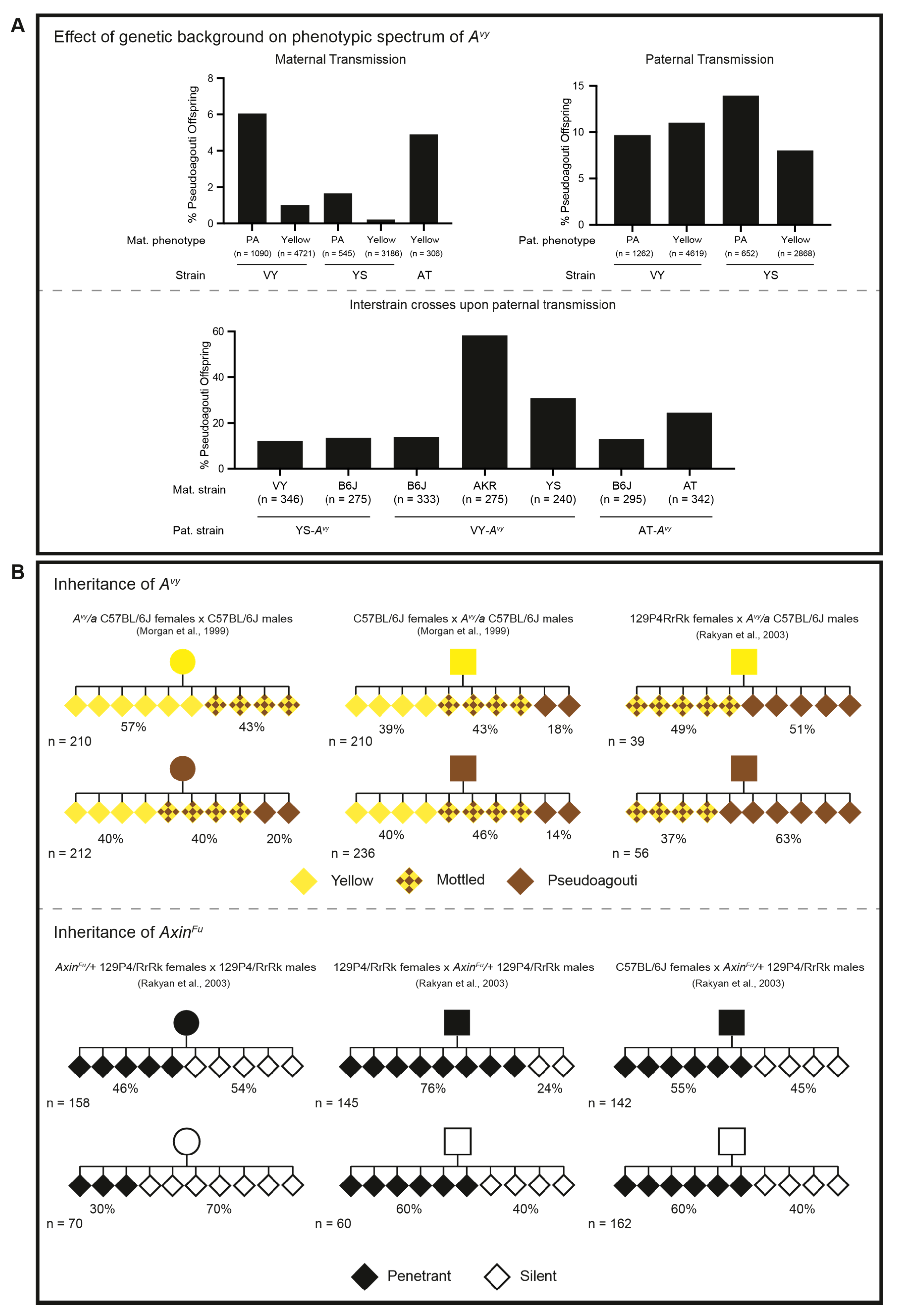
3.5. Epigenetic Inheritance of Metastable Epialleles, Avy and AxinFu
4. Lessons from Transgenes
5. KRAB-ZFPs as Effectors of Strain-Specific ERV Regulation
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Boeke, J.; Stoye, J. Retrotransposons, Endogenous Retroviruses, and the Evolution of Retroelements. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997; Volume 1, pp. 123–456. ISBN 0879695714. [Google Scholar]
- Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef] [PubMed]
- Jern, P.; Sperber, G.O.; Blomberg, J. Use of Endogenous Retroviral Sequences (ERVs) and structural markers for retroviral phylogenetic inference and taxonomy. Retrovirology 2005, 2, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gifford, R.; Kabat, P.; Martin, J.; Lynch, C.; Tristem, M. Evolution and Distribution of Class II-Related Endogenous Retroviruses. J. Virol. 2005, 79, 6478–6486. [Google Scholar] [CrossRef] [Green Version]
- Stocking, C.; Kozak, C.A. Endogenous retroviruses: Murine endogenous retroviruses. Cell. Mol. Life Sci. 2008, 65, 3383–3398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gifford, R.J.; Blomberg, J.; Coffin, J.M.; Fan, H.; Heidmann, T.; Mayer, J.; Stoye, J.; Tristem, M.; Johnson, W.E. Nomenclature for endogenous retrovirus (ERV) loci. Retrovirology 2018, 15, 59. [Google Scholar] [CrossRef] [Green Version]
- Mietz, J.A.; Grossman, Z.; Lueders, K.K.; Kuff, E.L. Nucleotide sequence of a complete mouse intracisternal A-particle genome: Relationship to known aspects of particle assembly and function. J. Virol. 1987, 61, 3020–3029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, P.J.; Macfarlan, T.S.; Lorincz, M.C. Long Terminal Repeats: From Parasitic Elements to Building Blocks of the Transcriptional Regulatory Repertoire. Mol. Cell 2016, 62, 766–776. [Google Scholar] [CrossRef] [Green Version]
- Gagnier, L.; Belancio, V.P.; Mager, D.L. Mouse germ line mutations due to retrotransposon insertions. Mob. DNA 2019, 10, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Ribet, D.; Dewannieux, M.; Heidmann, T. An active murine transposon family pair: Retrotransposition of “master” MusD copies and ETn trans-mobilization. Genome Res. 2004, 14, 2261–2267. [Google Scholar] [CrossRef] [Green Version]
- Maksakova, I.A.; Romanish, M.T.; Gagnier, L.; Dunn, C.A.; van de Lagemaat, L.N.; Mager, D.L. Retroviral Elements and Their Hosts: Insertional Mutagenesis in the Mouse Germ Line. PLoS Genet. 2006, 2, e2. [Google Scholar] [CrossRef] [Green Version]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements 06 Biological Sciences 0604 Genetics. Genome Biol. 2018, 19, 1–12. [Google Scholar] [CrossRef]
- Zhang, Y.; Maksakova, I.A.; Gagnier, L.; van de Lagemaat, L.N.; Mager, D.L. Genome-Wide Assessments Reveal Extremely High Levels of Polymorphism of Two Active Families of Mouse Endogenous Retroviral Elements. PLoS Genet. 2008, 4, e1000007. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Akagi, K.; Hu, Y.; Trivett, A.L.; Hlynialuk, C.J.W.; Swing, D.A.; Volfovsky, N.; Morgan, T.C.; Golubeva, Y.; Stephens, R.M.; et al. Mouse endogenous retroviruses can trigger premature transcriptional termination at a distance. Genome Res. 2012, 22, 870–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nellåker, C.; Keane, T.M.; Yalcin, B.; Wong, K.; Agam, A.; Belgard, T.G.; Flint, J.; Adams, D.J.; Frankel, W.N.; Ponting, C.P. The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol. 2012, 13, R45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thybert, D.; Roller, M.; Navarro, F.C.P.; Fiddes, I.; Streeter, I.; Feig, C.; Martin-Galvez, D.; Kolmogorov, M.; Janoušek, V.; Akanni, W.; et al. Repeat associated mechanisms of genome evolution and function revealed by the Mus caroli and Mus pahari genomes. Genome Res. 2018, 28, 448–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilue, J.; Doran, A.G.; Fiddes, I.T.; Abrudan, M.; Armstrong, J.; Bennett, R.; Chow, W.; Collins, J.; Collins, S.; Czechanski, A.; et al. Sixteen diverse laboratory mouse reference genomes define strain-specific haplotypes and novel functional loci. Nat. Genet. 2018, 50, 1574–1583. [Google Scholar] [CrossRef]
- Lilue, J.; Shivalikanjli, A.; Adams, D.J.; Keane, T.M. Mouse protein coding diversity: What’s left to discover? PLoS Genet. 2019, 15, e1008446. [Google Scholar] [CrossRef]
- Hamilton, B.A.; Yu, B.D. Modifier Genes and the Plasticity of Genetic Networks in Mice. PLoS Genet. 2012, 8, e1002644. [Google Scholar] [CrossRef] [Green Version]
- Perry, M.N.; Bello, S.M.; Smith, C.L. Know Your Model: Why mouse inbred strain contribution matters. Lab Anim. 2020, 49, 133–134. [Google Scholar] [CrossRef]
- Matsui, T.; Leung, D.; Miyashita, H.; Maksakova, I.A.; Miyachi, H.; Kimura, H.; Tachibana, M.; Lorincz, M.C.; Shinkai, Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 2010, 464, 927–931. [Google Scholar] [CrossRef] [Green Version]
- Karimi, M.M.; Goyal, P.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M.; et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mescs. Cell Stem Cell 2011, 8, 676–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, D.; Goff, S.P. Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 2009, 458, 1201–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, G.; Yang, P.; Füchtbauer, A.C.; Füchtbauer, E.M.; Silva, A.M.; Park, C.; Wu, W.; Nielsen, A.L.; Pedersen, F.S.; Macfarlan, T.S. The KRAB zinc finger protein ZFP809 is required to initiate epigenetic silencing of endogenous retroviruses. Genes Dev. 2015, 29, 538–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.-T. KAPtain in charge of multiple missions: Emerging roles of KAP1. World J. Biol. Chem. 2014, 5, 308. [Google Scholar] [CrossRef] [PubMed]
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7, 1–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ecco, G.; Imbeault, M.; Trono, D. KRAB zinc finger proteins. Development 2017, 144, 2719–2729. [Google Scholar] [CrossRef] [Green Version]
- Bruno, M.; Mahgoub, M.; Macfarlan, T.S. The Arms Race Between KRAB–Zinc Finger Proteins and Endogenous Retroelements and Its Impact on Mammals. Annu. Rev. Genet. 2019, 53, 393–416. [Google Scholar] [CrossRef]
- Rowe, H.M.; Jakobsson, J.; Mesnard, D.; Rougemont, J.; Reynard, S.; Aktas, T.; Maillard, P.V.; Layard-Liesching, H.; Verp, S.; Marquis, J.; et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 2010, 463, 237–240. [Google Scholar] [CrossRef]
- Liu, S.; Brind’Amour, J.; Karimi, M.M.; Shirane, K.; Bogutz, A.; Lefebvre, L.; Sasaki, H.; Shinkai, Y.; Lorincz, M.C. Setdb1 is required for germline development and silencing of H3K9me3-marked endogenous retroviruses in primordial germ cells. Genes Dev. 2014, 28, 2041–2055. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.P.; Chaillet, J.R.; Bestor, T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998, 20, 116. [Google Scholar] [CrossRef]
- Quenneville, S.; Turelli, P.; Bojkowska, K.; Raclot, C.; Offner, S.; Kapopoulou, A.; Trono, D. The KRAB-ZFP/KAP1 System Contributes to the Early Embryonic Establishment of Site-Specific DNA Methylation Patterns Maintained during Development. Cell Rep. 2012, 2, 766–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, H.M.; Friedli, M.; Offner, S.; Verp, S.; Mesnard, D.; Marquis, J.; Aktas, T.; Trono, D. De novo DNA methylation of endogenous retroviruses is shaped by KRAB-ZFPs/KAP1 and ESET. Development 2013, 140, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macfarlan, T.S.; Gifford, W.D.; Driscoll, S.; Lettieri, K.; Rowe, H.M.; Bonanomi, D.; Firth, A.; Singer, O.; Trono, D.; Pfaff, S.L. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 2012, 487, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Maksakova, I.A.; Thompson, P.J.; Goyal, P.; Jones, S.J.M.; Singh, P.B.; Karimi, M.M.; Lorincz, M.C. Distinct roles of KAP1, HP1 and G9a/GLP in silencing of the two-cell-specific retrotransposon MERVL in mouse ES cells. Epigenetics Chromatin 2013, 6, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macfarlan, T.S.; Gifford, W.D.; Agarwal, S.; Driscoll, S.; Lettieri, K.; Wang, J.; Andrews, S.E.; Franco, L.; Rosenfeld, M.G.; Ren, B. Endogenous retroviruses and neighboring genes are coordinately repressed by LSD1/KDM1A. Genes Dev. 2011, 25, 594–607. [Google Scholar] [CrossRef] [Green Version]
- Ancelin, K.; Syx, L.; Borensztein, M.; Ranisavljevic, N.; Vassilev, I.; Briseño-Roa, L.; Liu, T.; Metzger, E.; Servant, N.; Barillot, E.; et al. Maternal LSD1/KDM1A is an essential regulator of chromatin and transcription landscapes during zygotic genome activation. Elife 2016, 5. [Google Scholar] [CrossRef]
- Morgan, H.D.; Santos, F.; Green, K.; Dean, W.; Reik, W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005, 14, R47–R58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kigami, D.; Minami, N.; Takayama, H.; Imai, H. MuERV-L Is One of the Earliest Transcribed Genes in Mouse One-Cell Embryos1. Biol. Reprod. 2003, 68, 651–654. [Google Scholar] [CrossRef] [Green Version]
- Svoboda, P.; Stein, P.; Anger, M.; Bernstein, E.; Hannon, G.J.; Schultz, R.M. RNAi and expression of retrotransposons MuERV-L and IAP in preimplantation mouse embryos. Dev. Biol. 2004, 269, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Fu, B.; Ma, H.; Liu, D. Endogenous retroviruses function as gene expression regulatory elements during mammalian pre-implantation embryo development. Int. J. Mol. Sci. 2019, 20, 790. [Google Scholar] [CrossRef] [Green Version]
- Rowe, H.M.; Trono, D. Dynamic control of endogenous retroviruses during development. Virology 2011, 411, 273–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, D.C.; Lorincz, M.C. Silencing of endogenous retroviruses: When and why do histone marks predominate? Trends Biochem. Sci. 2012, 37, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Teissandier, A.; Pérez-Palacios, R.; Bourc’his, D. An epigenetic switch ensures transposon repression upon dynamic loss of DNA methylation in embryonic stem cells. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Sharif, J.; Endo, T.A.; Nakayama, M.; Xie, H.; Lorincz, M.C.; Correspondence, H.K. Activation of Endogenous Retroviruses in Dnmt1-/-ESCs Involves Disruption of SETDB1-Mediated Repression by NP95 Binding to Hemimethylated DNA Accession Numbers GSE77781 Sharif et al. Stem Cell 2016, 19, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Berrens, R.V.; Andrews, S.; Spensberger, D.; Santos, F.; Dean, W.; Gould, P.; Sharif, J.; Olova, N.; Chandra, T.; Koseki, H. An endosiRNA-based repression mechanism counteracts transposon activation during global DNA demethylation in embryonic stem cells. Cell Stem Cell 2017, 21, 694–703. [Google Scholar] [CrossRef] [Green Version]
- Schorn, A.J.; Martienssen, R. Tie-Break: Host and Retrotransposons Play tRNA. Trends Cell Biol. 2018, 28, 793–806. [Google Scholar] [CrossRef]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108. [Google Scholar] [CrossRef] [Green Version]
- Elsässer, S.J.; Noh, K.M.; Diaz, N.; Allis, C.D.; Banaszynski, L.A. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 2015, 522, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Voon, H.P.J.; Wong, L.H. New players in heterochromatin silencing: Histone variant H3. 3 and the ATRX/DAXX chaperone. Nucleic Acids Res. 2016, 44, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Wolf, G.; Rebollo, R.; Karimi, M.M.; Ewing, A.D.; Kamada, R.; Wu, W.; Wu, B.; Bachu, M.; Ozato, K.; Faulkner, G.J.; et al. On the role of H3.3 in retroviral silencing. Nature 2017, 548, E1–E3. [Google Scholar] [CrossRef]
- Hansen, J.C. Silencing the genome with linker histones. Proc. Natl. Acad. Sci. USA 2020, 202009513. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.K. Dactylaplasia in mice: A two-locus model for developmental anomalies. J. Hered. 1981, 72, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Sidow, A.; Bulotsky, M.S.; Kerrebrock, A.W.; Birren, B.W.; Altshuler, D.; Jaenisch, R.; Johnson, K.R.; Lander, E.S. A novel member of the F-box/WD40 gene family, encoding dactylin, is disrupted in the mouse dactylaplasia mutant. Nat. Genet. 1999, 23, 104–107. [Google Scholar] [CrossRef]
- Johnson, K.R.; Lane, P.W.; Ward-Bailey, P.; Davisson, M.T. Mapping the Mouse Dactylaplasia Mutation, Dac, and a Gene That Controls Its Expression, mdac. Genomics 1995, 29, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Kano, H.; Kurahashi, H.; Toda, T. Genetically regulated epigenetic transcriptional activation of retrotransposon insertion confers mouse dactylaplasia phenotype. Proc. Natl. Acad. Sci. USA 2007, 104, 19034–19039. [Google Scholar] [CrossRef] [Green Version]
- Friedli, M.; Nikolaev, S.; Lyle, R.; Arcangeli, M.; Duboule, D.; Spitz, F.; Antonarakis, S.E. Characterization of mouse Dactylaplasia mutations: A model for human ectrodactyly SHFM3. Mamm. Genome 2008, 19, 272–278. [Google Scholar] [CrossRef]
- Corsinotti, A.; Kapopoulou, A.; Gubelmann, C.; Imbeault, M.; Santoni de Sio, F.R.; Rowe, H.M.; Mouscaz, Y.; Deplancke, B.; Trono, D. Global and Stage Specific Patterns of Krüppel-Associated-Box Zinc Finger Protein Gene Expression in Murine Early Embryonic Cells. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Wolf, G.; de Iaco, A.; Sun, M.A.; Bruno, M.; Tinkham, M.; Hoang, D.; Mitra, A.; Ralls, S.; Trono, D.; Macfarlan, T.S. KRAB-zinc finger protein gene expansion in response to active retrotransposons in the murine lineage. Elife 2020, 9. [Google Scholar] [CrossRef]
- Juriloff, D.M. Differences in frequency of cleft lip among the a strains of mice. Teratology 1982, 25, 361–368. [Google Scholar] [CrossRef]
- Juriloff, D.M.; Harris, M.J.; Brown, C.J. Unravelling the complex genetics of cleft lip in the mouse model. Mamm. Genome 2001, 12, 426–435. [Google Scholar] [CrossRef]
- Juriloff, D.M.; Harris, M.J.; Dewell, S.L.; Brown, C.J.; Mager, D.L.; Gagnier, L.; Mah, D.G. Investigations of the genomic region that contains the clf1 mutation, a causal gene in multifactorial cleft lip and palate in mice. Birth Defects Res. Part A Clin. Mol. Teratol. 2005, 73, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J.; Mager, D.L.; Gagnier, L. Epigenetic Mechanism Causes Wnt9B Deficiency and Nonsyndromic Cleft Lip and Palate in the A/WySn Mouse Strain. Birth Defects Res. Part A Clin. Mol. Teratol. 2014, 100, 772–788. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.R.; Han, X.H.; Taketo, M.M.; Yoon, J.K. Wnt9b-dependent FGF signaling is crucial for outgrowth of the nasal and maxillary processes during upper jaw and lip development. Development 2012, 139, 1821–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juriloff, D.M.; Harris, M.J. Mouse genetic models of cleft lip with or without cleft palate. Birth Defects Res. Part A Clin. Mol. Teratol. 2008, 82, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Plamondon, J.A.; Harris, M.J.; Mager, D.L.; Gagnier, L.; Juriloff, D.M. The clf2 gene has an epigenetic role in the multifactorial etiology of cleft lip and palate in the A/WySn mouse strain. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Green, R.M.; Leach, C.L.; Diewert, V.M.; Aponte, J.D.; Schmidt, E.J.; Cheverud, J.M.; Roseman, C.C.; Young, N.M.; Marcucio, R.S.; Hallgrimsson, B. Nonlinear gene expression-phenotype relationships contribute to variation and clefting in the A/WySn mouse. Dev. Dyn. 2019, 248, 1232–1242. [Google Scholar] [CrossRef]
- Oliver, P.L.; Stoye, J.P. Genetic Analysis of Gv1, a Gene Controlling Transcription of Endogenous Murine Polytropic Proviruses. J. Virol. 1999, 73, 8227–8234. [Google Scholar] [CrossRef] [Green Version]
- Laporte, C.; Ballester, B.; Mary, C.; Izui, S.; Reininger, L. The Sgp3 Locus on Mouse Chromosome 13 Regulates Nephritogenic gp70 Autoantigen Expression and Predisposes to Autoimmunity. J. Immunol. 2003, 171, 3872–3877. [Google Scholar] [CrossRef] [Green Version]
- Treger, R.S.; Pope, S.D.; Kong, Y.; Tokuyama, M.; Taura, M.; Iwasaki, A. The Lupus Susceptibility Locus Sgp3 Encodes the Suppressor of Endogenous Retrovirus Expression SNERV. Immunity 2019, 50, 334–347.e9. [Google Scholar] [CrossRef] [Green Version]
- Politz, O.; Gratchev, A.; McCourt, P.A.G.; Schledzewski, K.; Guillot, P.; Johansson, S.; Svineng, G.; Franke, P.; Kannicht, C.; Kzhyshkowska, J.; et al. Stabilin-1 and -2 constitute a novel family of fasciclin-like hyaluronan receptor homologues. Biochem. J. 2002, 362, 155–164. [Google Scholar] [CrossRef]
- Harris, E.N.; Weigel, J.A.; Weigel, P.H. The human hyaluronan receptor for endocytosis (HARE/stabilin-2) is a systemic clearance receptor for heparin. J. Biol. Chem. 2008, 283, 17341–17350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schledzewski, K.; Géraud, C.; Arnold, B.; Wang, S.; Gröne, H.J.; Kempf, T.; Wollert, K.C.; Straub, B.K.; Schirmacher, P.; Demory, A.; et al. Deficiency of liver sinusoidal scavenger receptors stabilin-1 and -2 in mice causes glomerulofibrotic nephropathy via impaired hepatic clearance of noxious blood factors. J. Clin. Investig. 2011, 121, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Hirose, Y.; Saijou, E.; Sugano, Y.; Takeshita, F.; Nishimura, S.; Nonaka, H.; Chen, Y.R.; Sekine, K.; Kido, T.; Nakamura, T.; et al. Inhibition of Stabilin-2 elevates circulating hyaluronic acid levels and prevents tumor metastasis. Proc. Natl. Acad. Sci. USA 2012, 109, 4263–4268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayashima, Y.; Makhanova, N.A.; Matsuki, K.; Tomita, H.; Bennett, B.J.; Maeda, N. Identification of Aortic Arch-Specific Quantitative Trait Loci for Atherosclerosis by an Intercross of DBA/2J and 129S6 Apolipoprotein E-Deficient Mice. PLoS ONE 2015, 10, e0117478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda-Smithies, N.; Hiller, S.; Dong, S.; Kim, H.S.; Bennett, B.J.; Kayashima, Y. Ectopic expression of the Stabilin2 gene triggered by an intracisternal A particle (IAP) element in DBA/2J strain of mice. Mamm. Genome 2020, 31, 2–16. [Google Scholar] [CrossRef] [Green Version]
- Rakyan, V.K.; Blewitt, M.E.; Druker, R.; Preis, J.I.; Whitelaw, E. Metastable epialleles in mammals. Trends Genet. 2002, 18, 348–351. [Google Scholar] [CrossRef]
- Bertozzi, T.M.; Ferguson-Smith, A.C. Metastable epialleles and their contribution to epigenetic inheritance in mammals. Semin. Cell Dev. Biol. 2020, 97, 93–105. [Google Scholar] [CrossRef]
- Dickies, M.M. A New Viable Yellow Mutation in the House Mouse. J. Hered. 1962, 53, 84–86. [Google Scholar] [CrossRef]
- Morgan, H.D.; Sutherland, H.G.E.; Martin, D.I.K.; Whitelaw, E. Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet. 1999, 23, 314–318. [Google Scholar] [CrossRef]
- Reed, S.C. The inheritance and expression of Fused, a new mutation in the house mouse. Genetics 1937, 22, 1. [Google Scholar]
- Vasicek, T.J.; Zeng, L.; Guan, X.J.; Zhang, T.; Costantini, F.; Tilghman, S.M. Two dominant mutations in the mouse fused gene are the result of transposon insertions. Genetics 1997, 147, 777–786. [Google Scholar]
- Rakyan, V.K.; Chong, S.; Champ, M.E.; Cuthbert, P.C.; Morgan, H.D.; Luu, K.V.K.; Whitelaw, E. Transgenerational inheritance of epigenetic states at the murine AxinFu allele occurs after maternal and paternal transmission. Proc. Natl. Acad. Sci. USA 2003, 100, 2538–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duhl, D.M.J.; Vrieling, H.; Miller, K.A.; Wolff, G.L.; Barsh, G.S. Neomorphic agouti mutations in obese yellow mice. Nat. Genet. 1994, 8, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.L. Body composition and coat color correlation in different phenotypes of “viable yellow” mice. Science 1965, 147, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.L.; Roberts, D.W.; Mountjoy, K.G. Physiological consequences of ectopic agouti gene expression: The yellow obese mouse syndrome. Physiol. Genomics 1999, 1, 151–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, G.L. Genetic Modification of Homeostatic Regulation in the Mouse. Am. Nat. 1971, 105, 241–252. [Google Scholar] [CrossRef]
- Wolff, G.L.; Pitot, H.C. Variation of Hepatic Malic Enzyme Capacity with Hepatoma Susceptibility in Mice of Different Genotypes. Cancer Res. 1972, 32, 1861–1863. [Google Scholar]
- Wolff, G.L. Influence of maternal phenotype on metabolic differentiation of Agouti locus mutants in the mouse. Genetics 1978, 88, 529–539. [Google Scholar]
- Wolff, G.L.; Kodell, R.L.; Moore, S.R.; Cooney, C.A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998, 12, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Cooney, C.A.; Dave, A.A.; Wolff, G.L. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J. Nutr. 2002, 132, 2393S–2400S. [Google Scholar] [CrossRef] [Green Version]
- Belyaev, D.K.; Ruvinsky, A.O.; Borodin, P.M. Inheritance of alternative states of the fused gene in mice. J. Hered. 1981, 72, 107–112. [Google Scholar] [CrossRef]
- Ruvinsky, A.O.; Agulnik, A.I. Gametic imprinting and the manifestation of the fused gene in the house mouse. Dev. Genet. 1990, 11, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Oswald, J.; Engemann, S.; Lane, N.; Mayer, W.; Olek, A.; Fundele, R.; Dean, W.; Reik, W.; Walter, J. Active demethylation of the paternal genome in the mouse zygote. Curr. Biol. 2000, 10, 475–478. [Google Scholar] [CrossRef] [Green Version]
- Mayer, W.; Niveleau, A.; Walter, J.; Fundele, R.; Haaf, T. Embryogenesis: Demethylation of the zygotic paternal genome. Nature 2000, 403, 501–502. [Google Scholar] [CrossRef]
- Blewitt, M.E.; Vickaryous, N.K.; Paldi, A.; Koseki, H.; Whitelaw, E. Dynamic Reprogramming of DNA Methylation at an Epigenetically Sensitive Allele in Mice. PLoS Genet. 2006, 2, e49. [Google Scholar] [CrossRef] [PubMed]
- Kazachenka, A.; Bertozzi, T.M.; Sjoberg-Herrera, M.K.; Adams, S.; Adams, D.; Ferguson-Smith, A.C. Identification, Characterization, and Heritability of Murine Metastable Epialleles: Implications for Non-genetic Inheritance In Brief. Cell 2018, 175. [Google Scholar] [CrossRef] [Green Version]
- Bertozzi, T. Variable Methylation of Endogenous Retroviruses: Epigenetic Inheritance, Environmental Modulation, and Genetic Modifiers. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 2020. [Google Scholar] [CrossRef]
- Allen, N.D.; Norris, M.L.; Surani, M.A. Epigenetic control of transgene expression and imprinting by genotype-specific modifiers. Cell 1990, 61, 853–861. [Google Scholar] [CrossRef]
- Chaillet, J.R. Genomic imprinting: Lessons from mouse transgenes. Mutat. Res. 1994, 307, 441–449. [Google Scholar] [CrossRef]
- Daxinger, L.; Whitelaw, E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat. Rev. Genet. 2012, 13, 153–162. [Google Scholar] [CrossRef]
- Blewitt, M.; Whitelaw, E. The use of mouse models to study epigenetics. Cold Spring Harb. Perspect. Biol. 2013, 5, a017939. [Google Scholar] [CrossRef]
- Engler, P.; Storb, U. High-frequency deletional rearrangement of immunoglobulin kappa gene segments introduced into a pre-B-cell line. Proc. Natl. Acad. Sci. USA 1987, 84, 4949–4953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engler, P.; Roth, P.; Kim, J.Y.; Storb, U. Factors affecting the rearrangement efficiency of an Ig test gene. J. Immunol. 1991, 146, 2826–2835. [Google Scholar] [PubMed]
- Engler, P.; Haasch, D.; Pinkert, C.A.; Doglio, L.; Glymour, M.; Brinster, R.; Storb, U. A Strain-Specific Modifier on Mouse Chromosome 4 Controls the Methylation of Independent Transgene Loci. Cell 1991, 65, 939–947. [Google Scholar] [CrossRef]
- Ratnam, S.; Engler, P.; Bozek, G.; Mao, L.; Podlutsky, A.; Austad, S.; Martin, T.; Storb, U. Identification of Ssm1b, a novel modifier of DNA methylation, and its expression during mouse embryogenesis. Development 2014, 141, 2024–2034. [Google Scholar] [CrossRef] [Green Version]
- Weng, A.; Magnuson, T.; Storb, U. Strain-specific transgene methylation occurs early in mouse development and can be recapitulated in embryonic stem cells. Development 1995, 121, 2853–2859. [Google Scholar]
- Ratnam, S.; Bozek, G.; Martin, T.; Gallagher, S.J.; Payne, C.J.; Storb, U. Ssm1b expression and function in germ cells of adult mice and in early embryos. Mol. Reprod. Dev. 2017, 84, 596–613. [Google Scholar] [CrossRef]
- Kauzlaric, A.; Ecco, G.; Cassano, M.; Duc, J.; Imbeault, M.; Trono, D. The mouse genome displays highly dynamic populations of KRAB-zinc finger protein genes and related genetic units. PLoS ONE 2017, 12, e0173746. [Google Scholar] [CrossRef] [Green Version]
- Preis, J.I.; Downes, M.; Oates, N.A.; Rasko, J.E.J.; Whitelaw, E. Sensitive flow cytometric analysis reveals a novel type of parent-of-origin effect in the mouse genome. Curr. Biol. 2003, 13, 955–959. [Google Scholar] [CrossRef] [Green Version]
- Blewitt, M.E.; Vickaryous, N.K.; Hemley, S.J.; Ashe, A.; Bruxner, T.J.; Preis, J.I.; Arkell, R.; Whitelaw, E. An N-ethyl-N-nitrosourea screen for genes involved in variegation in the mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 7629–7634. [Google Scholar] [CrossRef] [Green Version]
- Chong, S.; Vickaryous, N.; Ashe, A.; Zamudio, N.; Youngson, N.; Hemley, S.; Stopka, T.; Skoultchi, A.; Matthews, J.; Scott, H.S.; et al. Modifiers of epigenetic reprogramming show paternal effects in the mouse. Nat. Genet. 2007, 39, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Ashe, A.; Morgan, D.K.; Whitelaw, N.C.; Bruxner, T.J.; Vickaryous, N.K.; Cox, L.L.; Butterfield, N.C.; Wicking, C.; Blewitt, M.E.; Wilkins, S.J.; et al. A genome-wide screen for modifiers of transgene variegation identifies genes with critical roles in development. Genome Biol. 2008, 9, R182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blewitt, M.E.; Gendrel, A.V.; Pang, Z.; Sparrow, D.B.; Whitelaw, N.; Craig, J.M.; Apedaile, A.; Hilton, D.J.; Dunwoodie, S.L.; Brockdorff, N.; et al. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat. Genet. 2008, 40, 663–669. [Google Scholar] [CrossRef]
- Daxinger, L.; Oey, H.; Apedaile, A.; Sutton, J.; Ashe, A.; Whitelaw, E. A forward genetic screen identifies eukaryotic translation initiation factor 3, subunit h (eiF3h), as an enhancer of variegation in the mouse. G3 Genes Genomes Genet. 2012, 2, 1393–1396. [Google Scholar] [CrossRef] [Green Version]
- Daxinger, L.; Harten, S.K.; Oey, H.; Epp, T.; Isbel, L.; Huang, E.; Whitelaw, N.; Apedaile, A.; Sorolla, A.; Yong, J.; et al. An ENU mutagenesis screen identifies novel and known genes involved in epigenetic processes in the mouse. Genome Biol. 2013, 14, R96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, C.J.; Larkins, L.K.; Price, R.; Tullis, K.M.; Miller, R.D.; Robins, D.M. Regulator of sex-limitation (Rs1) encodes a pair of KRAB zinc-finger genes that control sexually dimorphic liver gene expression. Genes Dev. 2003, 17, 2664–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavenhagen, J.B.; Robins, D.M. An ancient provirus has imposed androgen regulation on the adjacent mouse sex-limited protein gene. Cell 1988, 55, 247–254. [Google Scholar] [CrossRef]
- Krebs, C.J.; Schultz, D.C.; Robins, D.M. The KRAB Zinc Finger Protein RSL1 Regulates Sex- and Tissue-Specific Promoter Methylation and Dynamic Hormone-Responsive Chromatin Configuration. Mol. Cell. Biol. 2012, 32, 3732–3742. [Google Scholar] [CrossRef] [Green Version]
- Bojkowska, K.; Aloisio, F.; Cassano, M.; Kapopoulou, A.; Santoni de Sio, F.; Zangger, N.; Offner, S.; Cartoni, C.; Thomas, C.; Quenneville, S.; et al. Liver-specific ablation of Krüppel-associated box-associated protein 1 in mice leads to male-predominant hepatosteatosis and development of liver adenoma. Hepatology 2012, 56, 1279–1290. [Google Scholar] [CrossRef] [Green Version]
- Krebs, C.J.; Larkins, L.K.; Khan, S.M.; Robins, D.M. Expansion and diversification of KRAB zinc-finger genes within a cluster including Regulator of sex-limitation 1 and 2. Genomics 2005, 85, 752–761. [Google Scholar] [CrossRef]
- Juriloff, D.M.; Harris, M.J.; Dewell, S.L. A digenic cause of cleft lip in A-strain mice and definition of candidate genes for the two loci. Birth Defects Res. Part A Clin. Mol. Teratol. 2004, 70, 509–518. [Google Scholar] [CrossRef]
- Emerson, R.O.; Thomas, J.H. Adaptive evolution in zinc finger transcription factors. PLoS Genet. 2009, 5. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.H.; Schneider, S. Coevolution of retroelements and tandem zinc finger genes. Genome Res. 2011, 21, 1800–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbeault, M.; Helleboid, P.Y.; Trono, D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 2017, 543, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Mekada, K.; Abe, K.; Murakami, A.; Nakamura, S.; Nakata, H.; Moriwaki, K.; Obata, Y.; Yoshiki, A. Genetic differences among C57BL/6 substrains. Exp. Anim. 2009, 58, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.M.; Greenaway, S.; White, J.K.; Fuchs, H.; Gailus-Durner, V.; Wells, S.; Sorg, T.; Wong, K.; Bedu, E.; Cartwright, E.J.; et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 2013, 14, R82. [Google Scholar] [CrossRef]
- Helleboid, P.; Heusel, M.; Duc, J.; Piot, C.; Thorball, C.W.; Coluccio, A.; Pontis, J.; Imbeault, M.; Turelli, P.; Aebersold, R.; et al. The interactome of KRAB zinc finger proteins reveals the evolutionary history of their functional diversification. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Ecco, G.; Cassano, M.; Kauzlaric, A.; Duc, J.; Coluccio, A.; Offner, S.; Imbeault, M.; Rowe, H.M.; Turelli, P.; Trono, D. Transposable Elements and Their KRAB-ZFP Controllers Regulate Gene Expression in Adult Tissues. Dev. Cell 2016, 36, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ito, M.; Zhou, F.; Youngson, N.; Zuo, X.; Leder, P.; Ferguson-Smith, A.C. A Maternal-Zygotic Effect Gene, Zfp57, Maintains Both Maternal and Paternal Imprints. Dev. Cell 2008, 15, 547–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strogantsev, R.; Krueger, F.; Yamazawa, K.; Shi, H.; Gould, P.; Goldman-Roberts, M.; McEwen, K.; Sun, B.; Pedersen, R.; Ferguson-Smith, A.C. Allele-specific binding of ZFP57 in the epigenetic regulation of imprinted and non-imprinted monoallelic expression. Genome Biol. 2015, 16, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, N.; Coluccio, A.; Thorball, C.W.; Planet, E.; Shi, H.; Offner, S.; Turelli, P.; Imbeault, M.; Ferguson-Smith, A.C.; Trono, D. ZNF445 is a primary regulator of genomic imprinting. Genes Dev. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Strogantsev, R.; Takahashi, N.; Kazachenka, A.; Lorincz, M.C.; Hemberger, M.; Ferguson-Smith, A.C. ZFP57 regulation of transposable elements and gene expression within and beyond imprinted domains. Epigenet. Chromatin 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanzey, E.; Mcnamara, T.F.; Apostolou, E.; Tahiliani, M.; Correspondence, M.S. A Susceptibility Locus on Chromosome 13 Profoundly Impacts the Stability of Genomic Imprinting in Mouse Pluripotent Stem Cells. Cell Rep. 2020, 30, 3597–3604.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disrupted Locus (ERV Type) | Documented Strains with ERV Mutation | Strain-Specific Phenotype | Dominant Modifier | Documented Permissive Strains | Documented Repressive Strains | Coordinates of Modifier Locus (mm10) | References |
|---|---|---|---|---|---|---|---|
| Dac1j/2j (MusD) | Dac1j—SM/Ckc Dac2j—MRL/MpJ | Dactylaplasia | Mdac | BALB/cJ, A/J, 129/J, SM/Ckc, LG/Ckc, | CBA/J, C3H/J, C57BL/6J, DBA/2J, AKR/J, SWR/J | chr13:56–65Mb | [54,55,56] |
| clf1 (IΔ1-type IAP) | A/HeJ, A/WySnJ, A/J | Cleft lip with palate | Clf2 | “A” strains | C57BL/6J | chr13:64.95–67.9Mb | [63,66,122] |
| NEERVs | - | NEERV dysregulation and lupus pathology | Snerv1 and 2 | C57BL/6N, 129S1/Sv, NZB | C57BL/6J | chr13:65.66–66.7Mb | [70] |
| Stab2-IAP (IΔ1-type IAP) | DBA/2J | Elevated plasma HA | Stab2-modifier | DBA/2J | C57BL/6J, 129S6/Sv | chr13: 59.7–73Mb | [76] |
| HRD transgene | - | Transgene no longer undergoes V-J recombination | Ssm1b | DBA/2J, C3H/HeJ, SJL/J, CBA/J, SM/J, AKR/J | C57BL/6J, FVB/NJ, C57L/J, LP/J, 129/SvJ, BALB/cJ, A/J | chr4: 147.4–147.9Mb | [108,109,111] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elmer, J.L.; Ferguson-Smith, A.C. Strain-Specific Epigenetic Regulation of Endogenous Retroviruses: The Role of Trans-Acting Modifiers. Viruses 2020, 12, 810. https://doi.org/10.3390/v12080810
Elmer JL, Ferguson-Smith AC. Strain-Specific Epigenetic Regulation of Endogenous Retroviruses: The Role of Trans-Acting Modifiers. Viruses. 2020; 12(8):810. https://doi.org/10.3390/v12080810
Chicago/Turabian StyleElmer, Jessica L., and Anne C. Ferguson-Smith. 2020. "Strain-Specific Epigenetic Regulation of Endogenous Retroviruses: The Role of Trans-Acting Modifiers" Viruses 12, no. 8: 810. https://doi.org/10.3390/v12080810
APA StyleElmer, J. L., & Ferguson-Smith, A. C. (2020). Strain-Specific Epigenetic Regulation of Endogenous Retroviruses: The Role of Trans-Acting Modifiers. Viruses, 12(8), 810. https://doi.org/10.3390/v12080810