Silencing and Transcriptional Regulation of Endogenous Retroviruses: An Overview



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Epigenetic Silencing of Murine ERVs
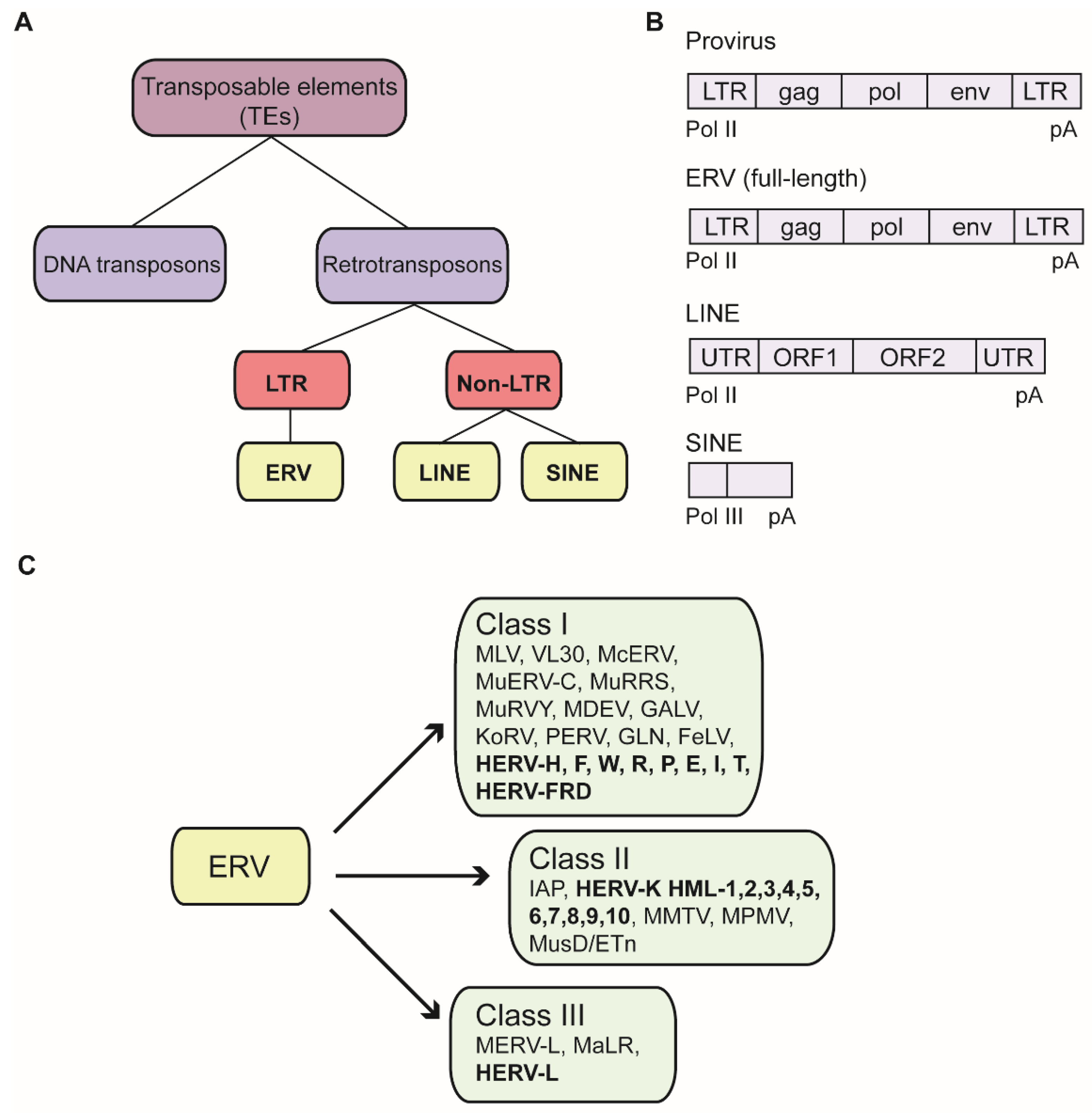
2.1. Nomenclature of Murine ERVs
2.2. Posttranslational Histone Modifications as a Driving Force in Heterochromatin Formation
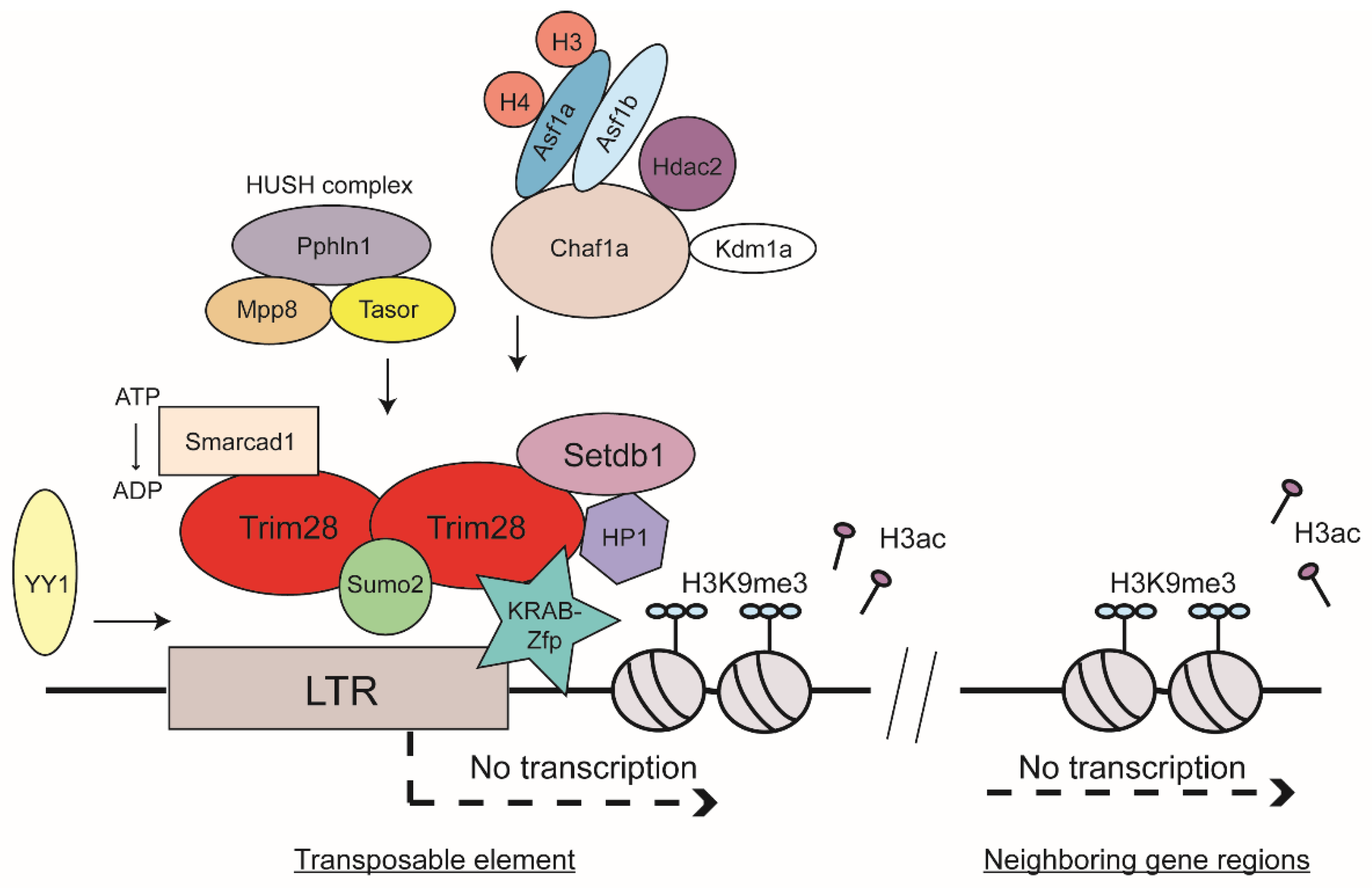
2.3. Trim28 as a Master Regulator of ERV Silencing
2.4. Sumoylation of Trim28 and HUSH Complex as Contributors to TE Silencing
2.5. Chromatin Spreading and ATP-Dependent Chromatin Remodeler
2.6. Histone Chaperones and Histone Variant H3.3 and Their Role in ERV Silencing
2.7. DNA Methylation as a Mechanism to Silence ERVs
2.8. RNA-Mediated Targeting of TEs
3. Transcriptional Regulation of Human ERVs
3.1. Nomenclature and Expression of Human ERVs
3.2. Transcriptional Regulation of Human ERVs
3.3. Deregulation of Human ERVs and Diseases
3.4. Human ERVs and Their Upregulation through Exogenous Viruses
4. Co-Option of ERV Functions for the Benefit of the Host
5. Concluding Remarks
Funding
Conflicts of Interest
References
- De Koning, A.P.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequencing, C.; Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvak, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef]
- Sultana, T.; Zamborlini, A.; Cristofari, G.; Lesage, P. Integration site selection by retroviruses and transposable elements in eukaryotes. Nat. Rev. Genet. 2017, 18, 292–308. [Google Scholar] [CrossRef] [PubMed]
- Groh, S.; Schotta, G. Silencing of endogenous retroviruses by heterochromatin. Cell Mol. Life Sci. 2017, 74, 2055–2065. [Google Scholar] [CrossRef]
- Bannert, N.; Kurth, R. The evolutionary dynamics of human endogenous retroviral families. Annu. Rev. Genomics Hum. Genet. 2006, 7, 149–173. [Google Scholar] [CrossRef]
- Johnson, W.E. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat. Rev. Microbiol. 2019, 17, 355–370. [Google Scholar] [CrossRef]
- McClintock, B. Controlling elements and the gene. Cold Spring Harb. Symp. Quant. Biol. 1956, 21, 197–216. [Google Scholar] [CrossRef]
- Baltimore, D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature 1970, 226, 1209–1211. [Google Scholar] [CrossRef]
- Temin, H.M.; Mizutani, S. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature 1970, 226, 1211–1213. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.N.; Carnegie, P.R.; Martin, J.; Davari Ejtehadi, H.; Hooley, P.; Roden, D.; Rowland-Jones, S.; Warren, P.; Astley, J.; Murray, P.G. Demystified ... Human endogenous retroviruses. Mol. Pathol. 2003, 56, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gifford, R.J.; Blomberg, J.; Coffin, J.M.; Fan, H.; Heidmann, T.; Mayer, J.; Stoye, J.; Tristem, M.; Johnson, W.E. Nomenclature for endogenous retrovirus (ERV) loci. Retrovirology 2018, 15, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupovic, M.; Blomberg, J.; Coffin, J.M.; Dasgupta, I.; Fan, H.; Geering, A.D.; Gifford, R.; Harrach, B.; Hull, R.; Johnson, W.; et al. Ortervirales: New virus order unifying five families of reverse-transcribing viruses. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.E. Endogenous retroviruses in the genomics era. Annu. Rev. Virol. 2015, 2, 135–159. [Google Scholar] [CrossRef]
- Stocking, C.; Kozak, C.A. Murine endogenous retroviruses. Cell Mol. Life Sci. 2008, 65, 3383–3398. [Google Scholar] [CrossRef] [Green Version]
- Rowe, W.P.; Lowy, D.R.; Teich, N.; Hartley, J.W. Some implications of the activation of murine leukemia virus by halogenated pyrimidines. Proc. Natl. Acad. Sci. USA 1972, 69, 1033–1035. [Google Scholar] [CrossRef] [Green Version]
- Maksakova, I.A.; Romanish, M.T.; Gagnier, L.; Dunn, C.A.; van de Lagemaat, L.N.; Mager, D.L. Retroviral elements and their hosts: Insertional mutagenesis in the mouse germ line. PLoS Genet. 2006, 2. [Google Scholar] [CrossRef] [Green Version]
- Boeke, J.D.; Stoye, J.P. Retrotransposons, endogenous retroviruses, and the evolution of retroelements. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor: New York, NY, USA, 1997. [Google Scholar]
- Callahan, R.; Smith, G.H. MMTV-induced mammary tumorigenesis: Gene discovery, progression to malignancy and cellular pathways. Oncogene 2000, 19, 992–1001. [Google Scholar] [CrossRef] [Green Version]
- Howard, G.; Eiges, R.; Gaudet, F.; Jaenisch, R.; Eden, A. Activation and transposition of endogenous retroviral elements in hypomethylation induced tumors in mice. Oncogene 2008, 27, 404–408. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, S.; Goff, S.P. Retroviral transcriptional regulation and embryonic stem cells: War and peace. Mol. Cell Biol. 2015, 35, 770–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grewal, S.I.; Jia, S. Heterochromatin revisited. Nat. Rev. Genet. 2007, 8, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Feuer, G.; Taketo, M.; Hanecak, R.C.; Fan, H. Two blocks in Moloney murine leukemia virus expression in undifferentiated F9 embryonal carcinoma cells as determined by transient expression assays. J. Virol. 1989, 63, 2317–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, O.; Yokota, Y.; Ishida, H.; Sugahara, T. Independent mechanisms involved in suppression of the Moloney leukemia virus genome during differentiation of murine teratocarcinoma cells. Cell 1983, 32, 1105–1113. [Google Scholar] [CrossRef]
- Teich, N.M.; Weiss, R.A.; Martin, G.R.; Lowy, D.R. Virus infection of murine teratocarcinoma stem cell lines. Cell 1977, 12, 973–982. [Google Scholar] [CrossRef]
- Lei, H.; Oh, S.P.; Okano, M.; Juttermann, R.; Goss, K.A.; Jaenisch, R.; Li, E. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development 1996, 122, 3195–3205. [Google Scholar]
- Machida, S.; Takizawa, Y.; Ishimaru, M.; Sugita, Y.; Sekine, S.; Nakayama, J.I.; Wolf, M.; Kurumizaka, H. Structural basis of heterochromatin formation by human HP1. Mol. Cell 2018, 69, 385–397. [Google Scholar] [CrossRef] [Green Version]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef]
- Day, D.S.; Luquette, L.J.; Park, P.J.; Kharchenko, P.V. Estimating enrichment of repetitive elements from high-throughput sequence data. Genome Biol. 2010, 11. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.Z.; Wang, Y.; Goff, S.P. Histones are rapidly loaded onto unintegrated retroviral DNAs soon after nuclear entry. Cell Host Microbe 2016, 20, 798–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Wang, G.Z.; Cingoz, O.; Goff, S.P. NP220 mediates silencing of unintegrated retroviral DNA. Nature 2018, 564, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Geis, F.K.; Goff, S.P. Unintegrated HIV-1 DNAs are loaded with core and linker histones and transcriptionally silenced. Proc. Natl. Acad. Sci. USA 2019, 116, 23735–23742. [Google Scholar] [CrossRef] [PubMed]
- Maksakova, I.A.; Mager, D.L.; Reiss, D. Keeping active endogenous retroviral-like elements in check: The epigenetic perspective. Cell Mol. Life Sci. 2008, 65, 3329–3347. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.C.; Dong, K.B.; Maksakova, I.A.; Goyal, P.; Appanah, R.; Lee, S.; Tachibana, M.; Shinkai, Y.; Lehnertz, B.; Mager, D.L.; et al. Lysine methyltransferase G9a is required for de novo DNA methylation and the establishment, but not the maintenance, of proviral silencing. Proc. Natl. Acad. Sci. USA 2011, 108, 5718–5723. [Google Scholar] [CrossRef] [Green Version]
- Maksakova, I.A.; Thompson, P.J.; Goyal, P.; Jones, S.J.; Singh, P.B.; Karimi, M.M.; Lorincz, M.C. Distinct roles of KAP1, HP1 and G9a/GLP in silencing of the two-cell-specific retrotransposon MERVL in mouse ES cells. Epigenetics Chromatin 2013, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Bulut-Karslioglu, A.; de La Rosa-Velazquez, I.A.; Ramirez, F.; Barenboim, M.; Onishi-Seebacher, M.; Arand, J.; Galan, C.; Winter, G.E.; Engist, B.; Gerle, B.; et al. Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol. Cell 2014, 55, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, K.; Shinkai, Y. SETDB1-Mediated silencing of retroelements. Viruses 2020, 12, 596. [Google Scholar] [CrossRef]
- Karimi, M.M.; Goyal, P.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M.; et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 2011, 8, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Leung, D.; Miyashita, H.; Maksakova, I.A.; Miyachi, H.; Kimura, H.; Tachibana, M.; Lorincz, M.C.; Shinkai, Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 2010, 464, 927–931. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Takemoto, K.; Shinkai, Y. A somatic role for the histone methyltransferase Setdb1 in endogenous retrovirus silencing. Nat. Commun. 2018, 9, 1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassi, D.A.; Jonsson, M.E.; Brattas, P.L.; Jakobsson, J. TRIM28 and the control of transposable elements in the brain. Brain Res. 2019, 1705, 43–47. [Google Scholar] [CrossRef]
- Rowe, H.M.; Jakobsson, J.; Mesnard, D.; Rougemont, J.; Reynard, S.; Aktas, T.; Maillard, P.V.; Layard-Liesching, H.; Verp, S.; Marquis, J.; et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 2010, 463, 237–240. [Google Scholar] [CrossRef]
- Cammas, F.; Mark, M.; Dolle, P.; Dierich, A.; Chambon, P.; Losson, R. Mice lacking the transcriptional corepressor TIF1beta are defective in early postimplantation development. Development 2000, 127, 2955–2963. [Google Scholar]
- Friedman, J.R.; Fredericks, W.J.; Jensen, D.E.; Speicher, D.W.; Huang, X.P.; Neilson, E.G.; Rauscher, F.J. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 1996, 10, 2067–2078. [Google Scholar] [CrossRef] [Green Version]
- Ecco, G.; Imbeault, M.; Trono, D. KRAB zinc finger proteins. Development 2017, 144, 2719–2729. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Wang, Y.; Macfarlan, T.S. The role of KRAB-ZFPs in transposable element repression and mammalian evolution. Trends Genet. 2017, 33, 871–881. [Google Scholar] [CrossRef]
- Emerson, R.O.; Thomas, J.H. Gypsy and the birth of the SCAN domain. J. Virol. 2011, 85, 12043–12052. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, D.; Stone, J.R.; Maki, J.L.; Collins, T.; Wagner, G. Mammalian SCAN domain dimer is a domain-swapped homolog of the HIV capsid C-terminal domain. Mol. Cell 2005, 17, 137–143. [Google Scholar] [CrossRef]
- Kingston, R.L.; Vogt, V.M. Domain swapping and retroviral assembly. Mol. Cell 2005, 17, 166–167. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G.; Greenberg, D.; Macfarlan, T.S. Spotting the enemy within: Targeted silencing of foreign DNA in mammalian genomes by the Kruppel-associated box zinc finger protein family. Mob. DNA 2015, 6, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, G.; de Iaco, A.; Sun, M.A.; Bruno, M.; Tinkham, M.; Hoang, D.; Mitra, A.; Ralls, S.; Trono, D.; Macfarlan, T.S. KRAB-zinc finger protein gene expansion in response to active retrotransposons in the murine lineage. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.; Mahgoub, M.; Macfarlan, T.S. The Arms Race Between KRAB-Zinc Finger Proteins and Endogenous Retroelements and Its Impact on Mammals. Annu. Rev. Genet. 2019, 53, 393–416. [Google Scholar] [CrossRef]
- Wolf, D.; Goff, S.P. Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 2009, 458, 1201–1204. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.; Goff, S.P. TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell 2007, 131, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Wolf, G.; Yang, P.; Fuchtbauer, A.C.; Fuchtbauer, E.M.; Silva, A.M.; Park, C.; Wu, W.; Nielsen, A.L.; Pedersen, F.S.; Macfarlan, T.S. The KRAB zinc finger protein ZFP809 is required to initiate epigenetic silencing of endogenous retroviruses. Genes Dev. 2015, 29, 538–554. [Google Scholar] [CrossRef] [Green Version]
- Treger, R.S.; Pope, S.D.; Kong, Y.; Tokuyama, M.; Taura, M.; Iwasaki, A. The lupus susceptibility locus sgp3 encodes the suppressor of endogenous retrovirus expression SNERV. Immunity 2019, 50, 334–347. [Google Scholar] [CrossRef] [Green Version]
- Seah, M.K.Y.; Wang, Y.; Goy, P.A.; Loh, H.M.; Peh, W.J.; Low, D.H.P.; Han, B.Y.; Wong, E.; Leong, E.L.; Wolf, G.; et al. The KRAB-zinc-finger protein ZFP708 mediates epigenetic repression at RMER19B retrotransposons. Development 2019, 146. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, S.; Lee, A.H.; Wang, G.Z.; Green, L.; Goff, S.P. Proviral silencing in embryonic cells is regulated by Yin Yang 1. Cell Rep. 2013, 4, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Coull, J.J.; Romerio, F.; Sun, J.M.; Volker, J.L.; Galvin, K.M.; Davie, J.R.; Shi, Y.; Hansen, U.; Margolis, D.M. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, J.R.; Becker, K.G.; Ennist, D.L.; Gleason, S.L.; Driggers, P.H.; Levi, B.Z.; Appella, E.; Ozato, K. Cloning of a negative transcription factor that binds to the upstream conserved region of Moloney murine leukemia virus. Mol. Cell Biol. 1992, 12, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satyamoorthy, K.; Park, K.; Atchison, M.L.; Howe, C.C. The intracisternal A-particle upstream element interacts with transcription factor YY1 to activate transcription: Pleiotropic effects of YY1 on distinct DNA promoter elements. Mol. Cell Biol. 1993, 13, 6621–6628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyde-DeRuyscher, R.P.; Jennings, E.; Shenk, T. DNA binding sites for the transcriptional activator/repressor YY1. Nucleic Acids Res. 1995, 23, 4457–4465. [Google Scholar] [CrossRef] [Green Version]
- Ryan, R.F.; Schultz, D.C.; Ayyanathan, K.; Singh, P.B.; Friedman, J.R.; Fredericks, W.J.; Rauscher, F.J. KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: A potential role for Kruppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol. Cell Biol. 1999, 19, 4366–4378. [Google Scholar] [CrossRef]
- Jacobs, S.A.; Taverna, S.D.; Zhang, Y.; Briggs, S.D.; Li, J.; Eissenberg, J.C.; Allis, C.D.; Khorasanizadeh, S. Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO J. 2001, 20, 5232–5241. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, S.A.; Khorasanizadeh, S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science 2002, 295, 2080–2083. [Google Scholar] [CrossRef]
- Lachner, M.; O’Carroll, D.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001, 410, 116–120. [Google Scholar] [CrossRef]
- Lechner, M.S.; Begg, G.E.; Speicher, D.W.; Rauscher, F.J. Molecular determinants for targeting heterochromatin protein 1-mediated gene silencing: Direct chromoshadow domain-KAP-1 corepressor interaction is essential. Mol. Cell Biol. 2000, 20, 6449–6465. [Google Scholar] [CrossRef]
- Brattas, P.L.; Jonsson, M.E.; Fasching, L.; Nelander, W.J.; Shahsavani, M.; Falk, R.; Falk, A.; Jern, P.; Parmar, M.; Jakobsson, J. TRIM28 controls a gene regulatory network based on endogenous retroviruses in human neural progenitor cells. Cell Rep. 2017, 18, 1–11. [Google Scholar] [CrossRef]
- Fasching, L.; Kapopoulou, A.; Sachdeva, R.; Petri, R.; Jonsson, M.E.; Manne, C.; Turelli, P.; Jern, P.; Cammas, F.; Trono, D.; et al. TRIM28 represses transcription of endogenous retroviruses in neural progenitor cells. Cell Rep. 2015, 10, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Boulard, M.; Rucli, S.; Edwards, J.R.; Bestor, T.H. Methylation-directed glycosylation of chromatin factors represses retrotransposon promoters. Proc. Natl. Acad. Sci. USA 2020, 117, 14292–14298. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.X.; El Farran, C.A.; Guo, H.C.; Yu, T.; Fang, H.T.; Wang, H.F.; Schlesinger, S.; Seah, Y.F.; Goh, G.Y.; Neo, S.P.; et al. Systematic identification of factors for provirus silencing in embryonic stem cells. Cell 2015, 163, 230–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.; Zhu, Y.; Sabo, Y.; Goff, S.P. Embryonic cells redistribute SUMO1 upon forced SUMO1 overexpression. mBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.V.; Peng, H.; Yurchenko, V.; Yap, K.L.; Negorev, D.G.; Schultz, D.C.; Psulkowski, E.; Fredericks, W.J.; White, D.E.; Maul, G.G.; et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 2007, 28, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Yap, K.L.; Ivanov, A.V.; Wang, X.; Mujtaba, S.; Plotnikova, O.; Rauscher, F.J.; Zhou, M.M. Structural insights into human KAP1 PHD finger-bromodomain and its role in gene silencing. Nat. Struct. Mol. Biol. 2008, 15, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Brummelkamp, T.R.; van Steensel, B. Gene regulation: A hush for transgene expression. Science 2015, 348, 1433–1434. [Google Scholar] [CrossRef]
- Tchasovnikarova, I.A.; Timms, R.T.; Matheson, N.J.; Wals, K.; Antrobus, R.; Gottgens, B.; Dougan, G.; Dawson, M.A.; Lehner, P.J. Gene silencing epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 2015, 348, 1481–1485. [Google Scholar] [CrossRef] [Green Version]
- Timms, R.T.; Tchasovnikarova, I.A.; Antrobus, R.; Dougan, G.; Lehner, P.J. ATF7IP-Mediated stabilization of the histone methyltransferase setdb1 is essential for heterochromatin formation by the HUSH complex. Cell Rep. 2016, 17, 653–659. [Google Scholar] [CrossRef] [Green Version]
- Tchasovnikarova, I.A.; Timms, R.T.; Douse, C.H.; Roberts, R.C.; Dougan, G.; Kingston, R.E.; Modis, Y.; Lehner, P.J. Hyperactivation of HUSH complex function by Charcot-Marie-Tooth disease mutation in MORC2. Nat. Genet. 2017, 49, 1035–1044. [Google Scholar] [CrossRef]
- Liu, N.; Lee, C.H.; Swigut, T.; Grow, E.; Gu, B.; Bassik, M.C.; Wysocka, J. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 2018, 553, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Robbez-Masson, L.; Tie, C.H.C.; Conde, L.; Tunbak, H.; Husovsky, C.; Tchasovnikarova, I.A.; Timms, R.T.; Herrero, J.; Lehner, P.J.; Rowe, H.M. The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes. Genome Res. 2018, 28, 836–845. [Google Scholar] [CrossRef] [Green Version]
- Groner, A.C.; Meylan, S.; Ciuffi, A.; Zangger, N.; Ambrosini, G.; Denervaud, N.; Bucher, P.; Trono, D. KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS Genet. 2010, 6, e1000869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebollo, R.; Karimi, M.M.; Bilenky, M.; Gagnier, L.; Miceli-Royer, K.; Zhang, Y.; Goyal, P.; Keane, T.M.; Jones, S.; Hirst, M.; et al. Retrotransposon-induced heterochromatin spreading in the mouse revealed by insertional polymorphisms. PLoS Genet. 2011, 7, e1002301. [Google Scholar] [CrossRef] [PubMed]
- Sachs, P.; Ding, D.; Bergmaier, P.; Lamp, B.; Schlagheck, C.; Finkernagel, F.; Nist, A.; Stiewe, T.; Mermoud, J.E. SMARCAD1 ATPase activity is required to silence endogenous retroviruses in embryonic stem cells. Nat. Commun. 2019, 10, 1335. [Google Scholar] [CrossRef] [Green Version]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef]
- Ding, D.; Bergmaier, P.; Sachs, P.; Klangwart, M.; Ruckert, T.; Bartels, N.; Demmers, J.; Dekker, M.; Poot, R.A.; Mermoud, J.E. The CUE1 domain of the SNF2-like chromatin remodeler SMARCAD1 mediates its association with KRAB-associated protein 1 (KAP1) and KAP1 target genes. J. Biol. Chem. 2018, 293, 2711–2724. [Google Scholar] [CrossRef] [Green Version]
- Macfarlan, T.S.; Gifford, W.D.; Agarwal, S.; Driscoll, S.; Lettieri, K.; Wang, J.; Andrews, S.E.; Franco, L.; Rosenfeld, M.G.; Ren, B.; et al. Endogenous retroviruses and neighboring genes are coordinately repressed by LSD1/KDM1A. Genes Dev. 2011, 25, 594–607. [Google Scholar] [CrossRef] [Green Version]
- Burgess, R.J.; Zhang, Z. Histones, histone chaperones and nucleosome assembly. Protein Cell 2010, 1, 607–612. [Google Scholar] [CrossRef] [Green Version]
- Thiru, A.; Nietlispach, D.; Mott, H.R.; Okuwaki, M.; Lyon, D.; Nielsen, P.R.; Hirshberg, M.; Verreault, A.; Murzina, N.V.; Laue, E.D. Structural basis of HP1/PXVXL motif peptide interactions and HP1 localisation to heterochromatin. EMBO J. 2004, 23, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Elsasser, S.J.; Noh, K.M.; Diaz, N.; Allis, C.D.; Banaszynski, L.A. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 2015, 522, 240–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, G.; Rebollo, R.; Karimi, M.M.; Ewing, A.D.; Kamada, R.; Wu, W.; Wu, B.; Bachu, M.; Ozato, K.; Faulkner, G.J.; et al. On the role of H3.3 in retroviral silencing. Nature 2017, 548. [Google Scholar] [CrossRef] [PubMed]
- Elsasser, S.J.; Noh, K.M.; Diaz, N.; Allis, C.D.; Banaszynski, L.A. Elsasser et al. reply. Nature 2017, 548. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [Green Version]
- Sadic, D.; Schmidt, K.; Groh, S.; Kondofersky, I.; Ellwart, J.; Fuchs, C.; Theis, F.J.; Schotta, G. Atrx promotes heterochromatin formation at retrotransposons. EMBO Rep. 2015, 16, 836–850. [Google Scholar] [CrossRef] [Green Version]
- Rowe, H.M.; Trono, D. Dynamic control of endogenous retroviruses during development. Virology 2011, 411, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Rowe, H.M.; Friedli, M.; Offner, S.; Verp, S.; Mesnard, D.; Marquis, J.; Aktas, T.; Trono, D. De novo DNA methylation of endogenous retroviruses is shaped by KRAB-ZFPs/KAP1 and ESET. Development 2013, 140, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Rose, N.R.; Klose, R.J. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta 2014, 1839, 1362–1372. [Google Scholar] [CrossRef] [Green Version]
- Gowher, H.; Liebert, K.; Hermann, A.; Xu, G.; Jeltsch, A. Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J. Biol. Chem. 2005, 280, 13341–13348. [Google Scholar] [CrossRef] [Green Version]
- Bourc’his, D.; Bestor, T.H. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 2004, 431, 96–99. [Google Scholar] [CrossRef]
- Sharif, J.; Endo, T.A.; Nakayama, M.; Karimi, M.M.; Shimada, M.; Katsuyama, K.; Goyal, P.; Brind’Amour, J.; Sun, M.A.; Sun, Z.; et al. Activation of endogenous retroviruses in Dnmt1(-/-) ESCs involves disruption of SETDB1-mediated repression by NP95 binding to hemimethylated DNA. Cell Stem Cell 2016, 19, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Min, B.; Park, J.S.; Jeong, Y.S.; Jeon, K.; Kang, Y.K. Dnmt1 binds and represses genomic retroelements via DNA methylation in mouse early embryos. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.P.; Chaillet, J.R.; Bestor, T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998, 20, 116–117. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.L.; Nishi, M.; Ohtsuka, T.; Matsui, T.; Takemoto, K.; Kamio-Miura, A.; Aburatani, H.; Shinkai, Y.; Kageyama, R. Essential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Development 2012, 139, 3806–3816. [Google Scholar] [CrossRef] [Green Version]
- Ohtani, H.; Liu, M.; Zhou, W.; Liang, G.; Jones, P.A. Switching roles for DNA and histone methylation depend on evolutionary ages of human endogenous retroviruses. Genome Res. 2018, 28, 1147–1157. [Google Scholar] [CrossRef] [Green Version]
- Stein, P.; Rozhkov, N.V.; Li, F.; Cardenas, F.L.; Davydenko, O.; Vandivier, L.E.; Gregory, B.D.; Hannon, G.J.; Schultz, R.M. Essential Role for endogenous siRNAs during meiosis in mouse oocytes. PLoS Genet. 2015, 11, e1005013. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Kannan, M.; Trivett, A.L.; Liao, H.; Wu, X.; Akagi, K.; Symer, D.E. An antisense promoter in mouse L1 retrotransposon open reading frame-1 initiates expression of diverse fusion transcripts and limits retrotransposition. Nucleic Acids Res. 2014, 42, 4546–4562. [Google Scholar] [CrossRef]
- Augui, S.; Nora, E.P.; Heard, E. Regulation of X-chromosome inactivation by the X-inactivation centre. Nat. Rev. Genet. 2011, 12, 429–442. [Google Scholar] [CrossRef]
- Zylicz, J.J.; Bousard, A.; Zumer, K.; Dossin, F.; Mohammad, E.; da Rocha, S.T.; Schwalb, B.; Syx, L.; Dingli, F.; Loew, D.; et al. The implication of early chromatin changes in X chromosome inactivation. Cell 2019, 176, 182–197.e23. [Google Scholar] [CrossRef] [Green Version]
- Minajigi, A.; Froberg, J.; Wei, C.; Sunwoo, H.; Kesner, B.; Colognori, D.; Lessing, D.; Payer, B.; Boukhali, M.; Haas, W.; et al. Chromosomes a comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, C.A.; Chen, C.K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Zhang, Q.C.; da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Magnuson, T.; Heard, E.; Chang, H.Y. Systematic discovery of Xist RNA binding proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Dossin, F.; Pinheiro, I.; Zylicz, J.J.; Roensch, J.; Collombet, S.; Le Saux, A.; Chelmicki, T.; Attia, M.; Kapoor, V.; Zhan, Y.; et al. SPEN integrates transcriptional and epigenetic control of X-inactivation. Nature 2020, 578, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.C.; Xu, J.; Nakamoto, M.Y.; Wei, Y.; Zarnegar, B.J.; Shi, Q.; Broughton, J.P.; Ransom, R.C.; Salhotra, A.; Nagaraja, S.D.; et al. Spen links RNA-mediated endogenous retrovirus silencing and X chromosome inactivation. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Seto, A.G.; Kingston, R.E.; Lau, N.C. The coming of age for Piwi proteins. Mol. Cell 2007, 26, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Pal-Bhadra, M.; Leibovitch, B.A.; Gandhi, S.G.; Chikka, M.R.; Bhadra, U.; Birchler, J.A.; Elgin, S.C. Heterochromatic silencing and HP1 localization in Drosophila are dependent on the RNAi machinery. Science 2004, 303, 669–672. [Google Scholar] [CrossRef] [Green Version]
- Aravin, A.A.; Sachidanandam, R.; Girard, A.; Fejes-Toth, K.; Hannon, G.J. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 2007, 316, 744–747. [Google Scholar] [CrossRef] [Green Version]
- Aravin, A.A.; Sachidanandam, R.; Bourc’his, D.; Schaefer, C.; Pezic, D.; Toth, K.F.; Bestor, T.; Hannon, G.J. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol. Cell 2008, 31, 785–799. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.N.; Bieniasz, P.D. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog. 2007, 3, e10. [Google Scholar] [CrossRef] [Green Version]
- Grabski, D.F.; Hu, Y.; Sharma, M.; Rasmussen, S.K. Close to the bedside: A systematic review of endogenous retroviruses and their impact in oncology. J. Surg. Res. 2019, 240, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Seifarth, W.; Frank, O.; Zeilfelder, U.; Spiess, B.; Greenwood, A.D.; Hehlmann, R.; Leib-Mosch, C. Comprehensive analysis of human endogenous retrovirus transcriptional activity in human tissues with a retrovirus-specific microarray. J. Virol. 2005, 79, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, I.K.; Rogozin, I.B.; Glazko, G.V.; Koonin, E.V. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. 2003, 19, 68–72. [Google Scholar] [CrossRef]
- Imbeault, M.; Helleboid, P.Y.; Trono, D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 2017, 543, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Najafabadi, H.S.; Mnaimneh, S.; Schmitges, F.W.; Garton, M.; Lam, K.N.; Yang, A.; Albu, M.; Weirauch, M.T.; Radovani, E.; Kim, P.M.; et al. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat. Biotechnol. 2015, 33, 555–562. [Google Scholar] [CrossRef]
- Schmitges, F.W.; Radovani, E.; Najafabadi, H.S.; Barazandeh, M.; Campitelli, L.F.; Yin, Y.; Jolma, A.; Zhong, G.; Guo, H.; Kanagalingam, T.; et al. Multiparameter functional diversity of human C2H2 zinc finger proteins. Genome Res. 2016, 26, 1742–1752. [Google Scholar] [CrossRef]
- Tie, C.H.; Fernandes, L.; Conde, L.; Robbez-Masson, L.; Sumner, R.P.; Peacock, T.; Rodriguez-Plata, M.T.; Mickute, G.; Gifford, R.; Towers, G.J.; et al. KAP1 regulates endogenous retroviruses in adult human cells and contributes to innate immune control. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Rajagopalan, D.; Tirado-Magallanes, R.; Bhatia, S.S.; Teo, W.S.; Sian, S.; Hora, S.; Lee, K.K.; Zhang, Y.; Jadhav, S.P.; Wu, Y.; et al. TIP60 represses activation of endogenous retroviral elements. Nucleic Acids Res. 2018, 46, 9456–9470. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Foroozesh, M.; Qin, Z. Transactivation of human endogenous retroviruses by tumor viruses and their functions in virus-associated malignancies. Oncogenesis 2019, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Pi, W.; Zhu, X.; Wu, M.; Wang, Y.; Fulzele, S.; Eroglu, A.; Ling, J.; Tuan, D. Long-range function of an intergenic retrotransposon. Proc. Natl. Acad. Sci. USA 2010, 107, 12992–12997. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, H.; Bachmann, S.; Schroder, J.; McArthur, J.; Torrey, E.F.; Yolken, R.H. Retroviral RNA identified in the cerebrospinal fluids and brains of individuals with schizophrenia. Proc. Natl. Acad. Sci. USA 2001, 98, 4634–4639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Lee, M.H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; von Geldern, G.; Johnson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Muotri, A.R.; Marchetto, M.C.; Coufal, N.G.; Oefner, R.; Yeo, G.; Nakashima, K.; Gage, F.H. L1 retrotransposition in neurons is modulated by MeCP2. Nature 2010, 468, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, E.; Arpino, C.; Matteucci, C.; Sorrentino, R.; Pica, F.; Alessandrelli, R.; Coniglio, A.; Curatolo, P.; Rezza, G.; Macciardi, F.; et al. HERVs expression in autism spectrum disorders. PLoS ONE 2012, 7, e48831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oluwole, S.O.; Yao, Y.; Conradi, S.; Kristensson, K.; Karlsson, H. Elevated levels of transcripts encoding a human retroviral envelope protein (syncytin) in muscles from patients with motor neuron disease. Amyotroph. Lateral. Scler. 2007, 8, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Douville, R.; Liu, J.; Rothstein, J.; Nath, A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann. Neurol. 2011, 69, 141–151. [Google Scholar] [CrossRef]
- Meyer, T.J.; Rosenkrantz, J.L.; Carbone, L.; Chavez, S.L. Endogenous retroviruses: With us and against us. Front. Chem. 2017, 5, 23. [Google Scholar] [CrossRef]
- van Horssen, J.; van der Pol, S.; Nijland, P.; Amor, S.; Perron, H. Human endogenous retrovirus W in brain lesions: Rationale for targeted therapy in multiple sclerosis. Mult. Scler. Relat. Disord. 2016, 8, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Kremer, D.; Gruchot, J.; Weyers, V.; Oldemeier, L.; Gottle, P.; Healy, L.; Ho Jang, J.; Kang, T.X.Y.; Volsko, C.; Dutta, R.; et al. pHERV-W envelope protein fuels microglial cell-dependent damage of myelinated axons in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 15216–15225. [Google Scholar] [CrossRef] [Green Version]
- Ruprecht, K.; Mayer, J. On the origin of a pathogenic HERV-W envelope protein present in multiple sclerosis lesions. Proc. Natl. Acad. Sci. USA 2019, 116, 19791–19792. [Google Scholar] [CrossRef] [Green Version]
- Balestrieri, E.; Cipriani, C.; Matteucci, C.; Benvenuto, A.; Coniglio, A.; Argaw-Denboba, A.; Toschi, N.; Bucci, I.; Miele, M.T.; Grelli, S.; et al. Children with autism spectrum disorder and their mothers share abnormal expression of selected endogenous retroviruses families and cytokines. Front. Immunol. 2019, 10, 2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payer, L.M.; Burns, K.H. Transposable elements in human genetic disease. Nat. Rev. Genet. 2019, 20, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Tovo, P.A.; Garazzino, S.; Dapra, V.; Alliaudi, C.; Silvestro, E.; Calvi, C.; Montanari, P.; Galliano, I.; Bergallo, M. Chronic HCV infection is associated with overexpression of human endogenous retroviruses that persists after drug-induced viral clearance. Int. J. Mol. Sci. 2020, 21, 3980. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hernandez, M.J.; Swanson, M.D.; Contreras-Galindo, R.; Cookinham, S.; King, S.R.; Noel, R.J., Jr.; Kaplan, M.H.; Markovitz, D.M. Expression of human endogenous retrovirus type K (HML-2) is activated by the Tat protein of HIV-1. J. Virol. 2012, 86, 7790–7805. [Google Scholar] [CrossRef] [Green Version]
- Zeilfelder, U.; Frank, O.; Sparacio, S.; Schon, U.; Bosch, V.; Seifarth, W.; Leib-Mosch, C. The potential of retroviral vectors to cotransfer human endogenous retroviruses (HERVs) from human packaging cell lines. Gene 2007, 390, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Mangeney, M.; de Parseval, N.; Thomas, G.; Heidmann, T. The full-length envelope of an HERV-H human endogenous retrovirus has immunosuppressive properties. J. Gen. Virol. 2001, 82, 2515–2518. [Google Scholar] [CrossRef] [Green Version]
- Lemaitre, C.; Tsang, J.; Bireau, C.; Heidmann, T.; Dewannieux, M. A human endogenous retrovirus-derived gene that can contribute to oncogenesis by activating the ERK pathway and inducing migration and invasion. PLoS Pathog. 2017, 13, e1006451. [Google Scholar] [CrossRef]
- Robbez-Masson, L.; Rowe, H.M. Retrotransposons shape species-specific embryonic stem cell gene expression. Retrovirology 2015, 12, 45. [Google Scholar] [CrossRef] [Green Version]
- Gifford, W.D.; Pfaff, S.L.; Macfarlan, T.S. Transposable elements as genetic regulatory substrates in early development. Trends Cell Biol. 2013, 23, 218–226. [Google Scholar] [CrossRef] [Green Version]
- Rebollo, R.; Romanish, M.T.; Mager, D.L. Transposable elements: An abundant and natural source of regulatory sequences for host genes. Annu. Rev. Genet. 2012, 46, 21–42. [Google Scholar] [CrossRef]
- Jern, P.; Coffin, J.M. Effects of retroviruses on host genome function. Annu. Rev. Genet. 2008, 42, 709–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, M.; Goudot, C.; Hoyler, T.; Lemoine, B.; Amigorena, S.; Zueva, E. Specific subfamilies of transposable elements contribute to different domains of T lymphocyte enhancers. Proc. Natl. Acad. Sci. USA 2020, 117, 7905–7916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Han, G.Z. Frequent retroviral gene co-option during the evolution of vertebrates. Mol. Biol. Evol. 2020. [Google Scholar] [CrossRef]
- Wang, T.; Zeng, J.; Lowe, C.B.; Sellers, R.G.; Salama, S.R.; Yang, M.; Burgess, S.M.; Brachmann, R.K.; Haussler, D. Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc. Natl. Acad. Sci. USA 2007, 104, 18613–18618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef] [Green Version]
- Manghera, M.; Douville, R.N. Endogenous retrovirus-K promoter: A landing strip for inflammatory transcription factors? Retrovirology 2013, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manghera, M.; Ferguson-Parry, J.; Lin, R.; Douville, R.N. NF-kappaB and IRF1 induce endogenous retrovirus k expression via interferon-stimulated response elements in Its 5′ long terminal repeat. J. Virol. 2016, 90, 9338–9349. [Google Scholar] [CrossRef] [Green Version]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef]
- Grow, E.J.; Flynn, R.A.; Chavez, S.L.; Bayless, N.L.; Wossidlo, M.; Wesche, D.J.; Martin, L.; Ware, C.B.; Blish, C.A.; Chang, H.Y.; et al. Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature 2015, 522, 221–225. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Sachs, F.; Ramsay, L.; Jacques, P.E.; Goke, J.; Bourque, G.; Ng, H.H. The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nat. Struct. Mol. Biol. 2014, 21, 423–425. [Google Scholar] [CrossRef]
- Fort, A.; Hashimoto, K.; Yamada, D.; Salimullah, M.; Keya, C.A.; Saxena, A.; Bonetti, A.; Voineagu, I.; Bertin, N.; Kratz, A.; et al. Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat. Genet. 2014, 46, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Santoni, F.A.; Guerra, J.; Luban, J. HERV-H RNA is abundant in human embryonic stem cells and a precise marker for pluripotency. Retrovirology 2012, 9, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theunissen, T.W.; Friedli, M.; He, Y.; Planet, E.; O’Neil, R.C.; Markoulaki, S.; Pontis, J.; Wang, H.; Iouranova, A.; Imbeault, M.; et al. Molecular criteria for defining the naive human pluripotent state. Cell Stem Cell 2016, 19, 502–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, T.; Preissl, S.; Amaral, M.L.; Grinstein, J.D.; Farah, E.N.; Destici, E.; Qiu, Y.; Hu, R.; Lee, A.Y.; et al. Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat. Genet. 2019, 51, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.H.; Saha, K.; Jaenisch, R. Pluripotency and cellular reprogramming: Facts, hypotheses, unresolved issues. Cell 2010, 143, 508–525. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, N.V.; Loewer, S.; Daley, G.Q.; Izsvak, Z.; Lower, J.; Lower, R. Human endogenous retrovirus K (HML-2) RNA and protein expression is a marker for human embryonic and induced pluripotent stem cells. Retrovirology 2013, 10, 115. [Google Scholar] [CrossRef] [Green Version]
- Ecco, G.; Cassano, M.; Kauzlaric, A.; Duc, J.; Coluccio, A.; Offner, S.; Imbeault, M.; Rowe, H.M.; Turelli, P.; Trono, D. Transposable elements and their KRAB-ZFP controllers regulate gene expression in adult tissues. Dev. Cell 2016, 36, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Wang, Y.; Hoang, D.; Tinkham, M.; Patel, A.; Sun, M.A.; Wolf, G.; Baker, M.; Chien, H.C.; Lai, K.N.; et al. A placental growth factor is silenced in mouse embryos by the zinc finger protein ZFP568. Science 2017, 356, 757–759. [Google Scholar] [CrossRef] [Green Version]
- Rote, N.S.; Chakrabarti, S.; Stetzer, B.P. The role of human endogenous retroviruses in trophoblast differentiation and placental development. Placenta 2004, 25, 673–683. [Google Scholar] [CrossRef]
- Grandi, N.; Tramontano, E. HERV Envelope proteins: Physiological role and pathogenic potential in cancer and autoimmunity. Front. Microbiol. 2018, 9, 462. [Google Scholar] [CrossRef]
- Ikeda, H.; Odaka, T. A cell membrane ‘gp70’ associated with Fv-4 gene: Immunological characterization, and tissue and strain distribution. Virology 1984, 133, 65–76. [Google Scholar] [CrossRef]
- Inaguma, Y.; Yoshida, T.; Ikeda, H. Scheme for the generation of a truncated endogenous murine leukaemia virus, the Fv-4 resistance gene. J. Gen. Virol. 1992, 73, 1925–1930. [Google Scholar] [CrossRef] [PubMed]
- Benit, L.; de Parseval, N.; Casella, J.F.; Callebaut, I.; Cordonnier, A.; Heidmann, T. Cloning of a new murine endogenous retrovirus, MuERV-L, with strong similarity to the human HERV-L element and with a gag coding sequence closely related to the Fv1 restriction gene. J. Virol. 1997, 71, 5652–5657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goff, S.P. Retrovirus restriction factors. Mol. Cell 2004, 16, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Best, S.; Le Tissier, P.; Towers, G.; Stoye, J.P. Positional cloning of the mouse retrovirus restriction gene Fv1. Nature 1996, 382, 826–829. [Google Scholar] [CrossRef] [PubMed]
- Cottee, M.A.; Letham, S.C.; Young, G.R.; Stoye, J.P.; Taylor, I.A. Structure of Drosophila melanogaster ARC1 reveals a repurposed molecule with characteristics of retroviral Gag. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastuzyn, E.D.; Day, C.E.; Kearns, R.B.; Kyrke-Smith, M.; Taibi, A.V.; McCormick, J.; Yoder, N.; Belnap, D.M.; Erlendsson, S.; Morado, D.R.; et al. The neuronal gene arc encodes a repurposed retrotransposon gag protein that mediates intercellular RNA transfer. Cell 2018, 173, 275. [Google Scholar] [CrossRef] [Green Version]
- Ashley, J.; Cordy, B.; Lucia, D.; Fradkin, L.G.; Budnik, V.; Thomson, T. Retrovirus-like gag protein Arc1 binds RNA and traffics across synaptic boutons. Cell 2018, 172, 262–274. [Google Scholar] [CrossRef] [Green Version]
- Ishak, C.A.; Classon, M.; de Carvalho, D.D. Deregulation of retroelements as an emerging therapeutic opportunity in cancer. Trends Cancer 2018, 4, 583–597. [Google Scholar] [CrossRef]
- Roulois, D.; Loo, Y.H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [Green Version]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbot, W.; Dupressoir, A.; Lazar, V.; Heidmann, T. Epigenetic regulation of an IAP retrotransposon in the aging mouse: Progressive demethylation and de-silencing of the element by its repetitive induction. Nucleic Acids Res. 2002, 30, 2365–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.H.; Horiuchi, M.; Itoh, T.; Greenhalgh, D.G.; Cho, K. Cerebellum-specific and age-dependent expression of an endogenous retrovirus with intact coding potential. Retrovirology 2011, 8, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevalainen, T.; Autio, A.; Mishra, B.H.; Marttila, S.; Jylha, M.; Hurme, M. Aging-associated patterns in the expression of human endogenous retroviruses. PLoS ONE 2018, 13, e0207407. [Google Scholar] [CrossRef] [PubMed]
- Sankowski, R.; Strohl, J.J.; Huerta, T.S.; Nasiri, E.; Mazzarello, A.N.; D’Abramo, C.; Cheng, K.F.; Staszewski, O.; Prinz, M.; Huerta, P.T.; et al. Endogenous retroviruses are associated with hippocampus-based memory impairment. Proc. Natl. Acad. Sci. USA 2019, 116, 25982–25990. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geis, F.K.; Goff, S.P. Silencing and Transcriptional Regulation of Endogenous Retroviruses: An Overview. Viruses 2020, 12, 884. https://doi.org/10.3390/v12080884
Geis FK, Goff SP. Silencing and Transcriptional Regulation of Endogenous Retroviruses: An Overview. Viruses. 2020; 12(8):884. https://doi.org/10.3390/v12080884
Chicago/Turabian StyleGeis, Franziska K., and Stephen P. Goff. 2020. "Silencing and Transcriptional Regulation of Endogenous Retroviruses: An Overview" Viruses 12, no. 8: 884. https://doi.org/10.3390/v12080884
APA StyleGeis, F. K., & Goff, S. P. (2020). Silencing and Transcriptional Regulation of Endogenous Retroviruses: An Overview. Viruses, 12(8), 884. https://doi.org/10.3390/v12080884