Advances with RNAi-Based Therapy for Hepatitis B Virus Infection

 ,
,
Abstract
:1. Introduction
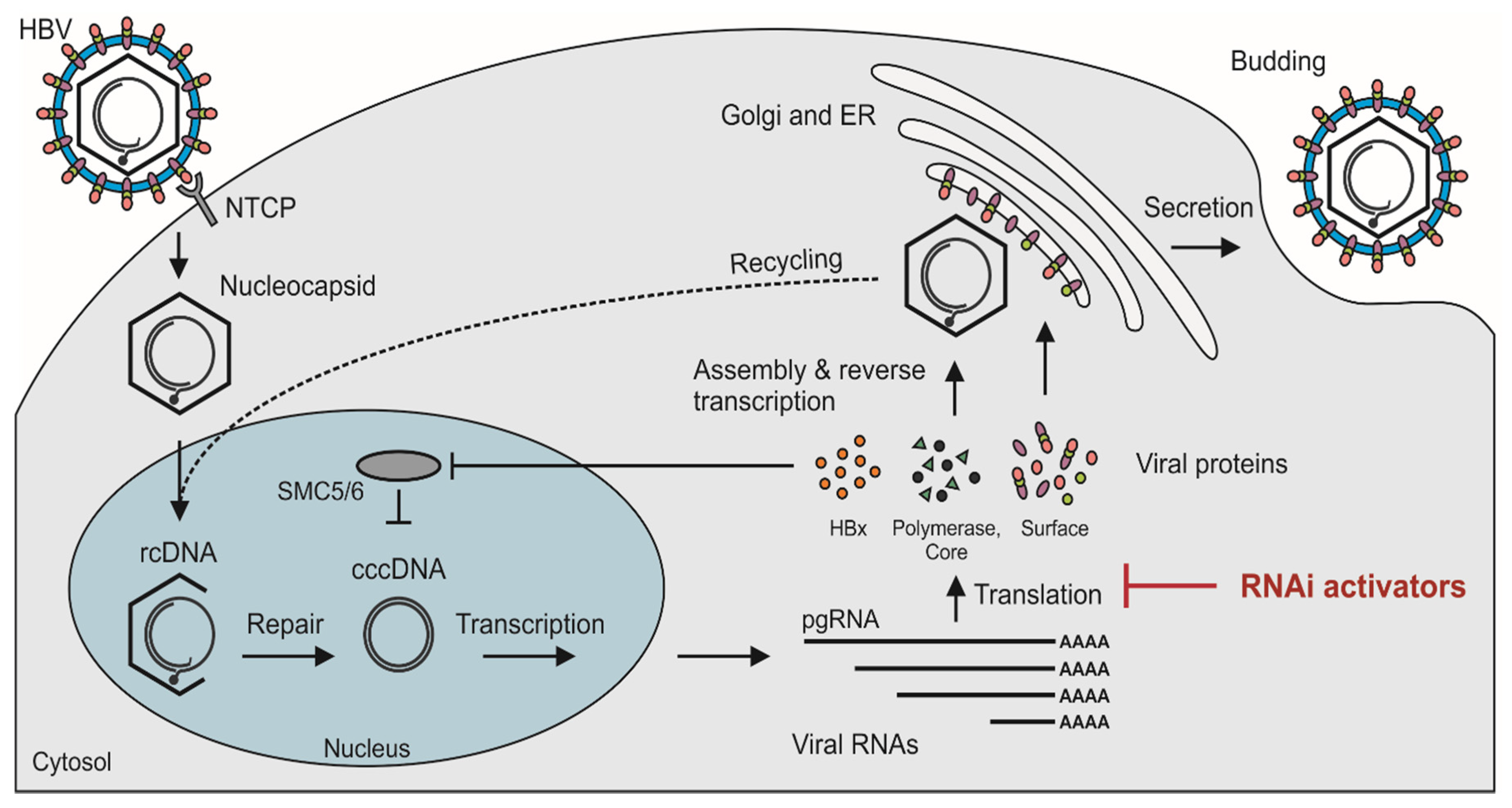
2. HBV Replication
3. Goals of Treating Chronic Infection with HBV
4. Currently Licensed Treatment for HBV
5. New HBV Drugs under Development
5.1. Rationale for Advancing RNAi-Based Anti-HBV Therapy
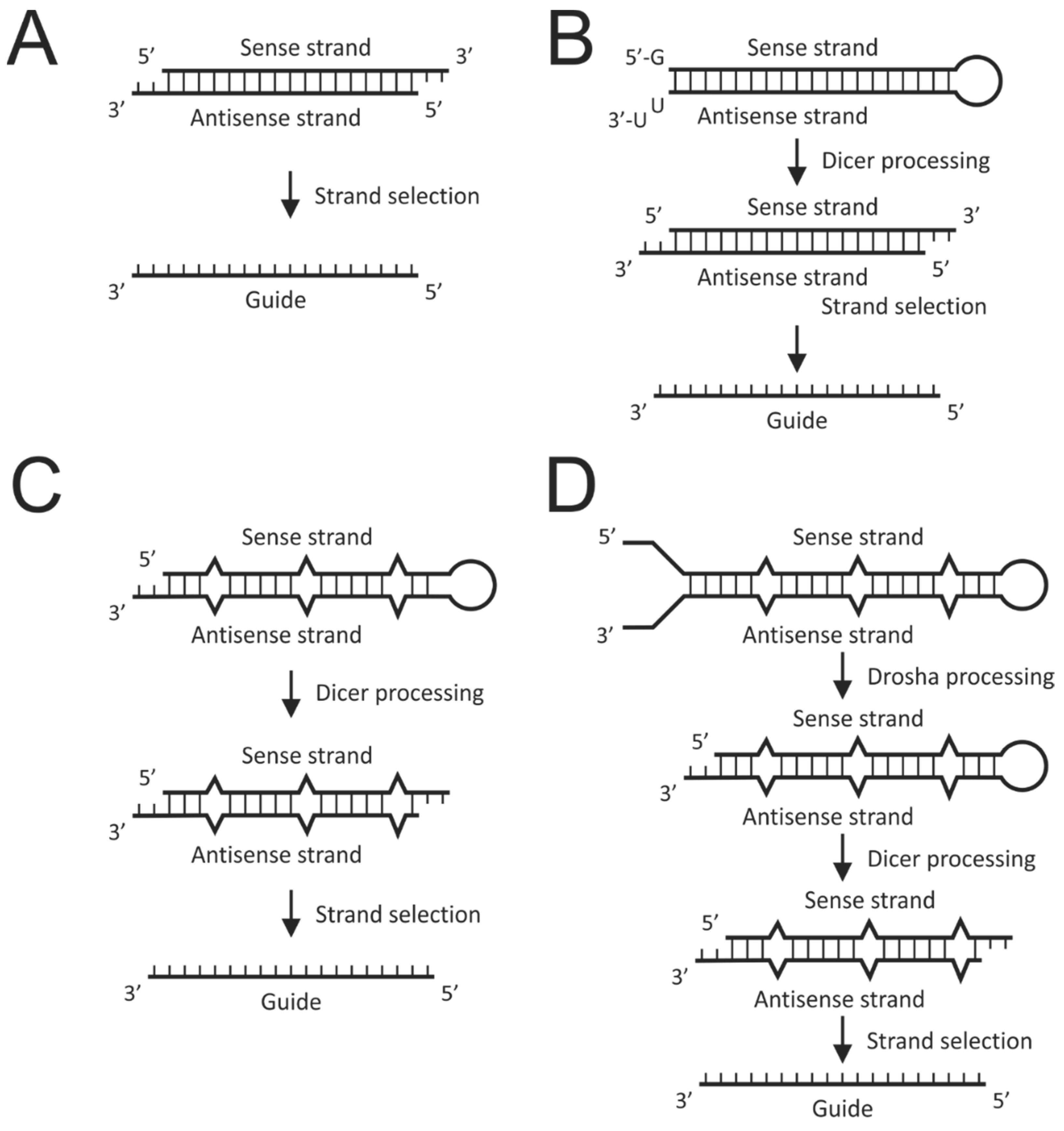
5.2. The RNAi Pathway
5.3. RNAi Activators
5.4. Significance of Genotype Variability for Advancing RNAi-Based HBV Therapy
6. Models of HBV Infection
7. Delivery of HBV-Targeting Gene Silencers
7.1. Non-Viral Vectors
7.2. Viral Vectors
7.3. Clinical Trials Evaluating RNAi-Based Treatment for HBV Infection
8. Future of the Field
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Revill, P.A.; Chisari, F.V.; Block, J.M.; Dandri, M.; Gehring, A.J.; Guo, H.; Hu, J.; Kramvis, A.; Lampertico, P.; Janssen, H.L.A.; et al. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol. Hepatol. 2019, 4, 545–558. [Google Scholar] [CrossRef]
- Huovila, A.P.; Eder, A.M.; Fuller, S.D. Hepatitis B surface antigen assembles in a post-ER, pre-Golgi compartment. J. Cell Biol. 1992, 118, 1305–1320. [Google Scholar] [CrossRef] [PubMed]
- Steven, A.C.; Conway, J.F.; Cheng, N.; Watts, N.R.; Belnap, D.M.; Harris, A.; Stahl, S.J.; Wingfield, P.T. Structure, assembly, and antigenicity of hepatitis B virus capsid proteins. Adv. Virus Res. 2005, 64, 125–164. [Google Scholar] [PubMed]
- Maupas, P.; Goudeau, A.; Coursaget, P.; Drucker, J.; Bagros, P. Immunisation against hepatitis B in man. Lancet 1976, 1, 1367–1370. [Google Scholar] [CrossRef]
- Purcell, R.H.; Gerin, J.L. Hepatitis B subunit vaccine: A preliminary report of safety and efficacy tests in chimpanzees. Am. J. Med. Sci. 1975, 270, 395–400. [Google Scholar] [CrossRef]
- Szmuness, W.; Stevens, C.E.; Harley, E.J.; Zang, E.A.; Oleszko, W.R.; William, D.C.; Kellner, A. Hepatitis B vaccine: Demonstration of efficacy in a controlled clinical trial in a high-risk population in the United States. N. Engl. J. Med. 1980, 303, 833–841. [Google Scholar] [CrossRef]
- Sureau, C.; Salisse, J. A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis B virus a-determinant. Hepatology 2013, 57, 985–994. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Koniger, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef]
- Iwamoto, M.; Saso, W.; Nishioka, K.; Ohashi, H.; Sugiyama, R.; Ryo, A.; Ohki, M.; Yun, J.H.; Park, S.Y.; Ohshima, T.; et al. The machinery for endocytosis of epidermal growth factor receptor coordinates the transport of incoming hepatitis B virus to the endosomal network. J. Biol. Chem. 2020, 295, 800–807. [Google Scholar] [CrossRef]
- Iwamoto, M.; Saso, W.; Sugiyama, R.; Ishii, K.; Ohki, M.; Nagamori, S.; Suzuki, R.; Aizaki, H.; Ryo, A.; Yun, J.H.; et al. Epidermal growth factor receptor is a host-entry cofactor triggering hepatitis B virus internalization. Proc. Natl. Acad. Sci. USA 2019, 116, 8487–8492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Ou, J.H. Phosphorylation and nuclear localization of the hepatitis B virus core protein: Significance of serine in the three repeated SPRRR motifs. J. Virol. 1995, 69, 1025–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koniger, C.; Wingert, I.; Marsmann, M.; Rösler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, K.; Que, L.; Shimadu, M.; Koura, M.; Ishihara, Y.; Wakae, K.; Nakamura, T.; Watashi, K.; Wakita, T.; Muramatsu, M. Flap endonuclease 1 is involved in cccDNA formation in the hepatitis B virus. PLoS Pathog. 2018, 14, e1007124. [Google Scholar] [CrossRef]
- Asabe, S.; Wieland, S.F.; Chattopadhyay, P.K.; Roederer, M.; Engle, R.E.; Purcell, R.H.; Chisari, F.V. The size of the viral inoculum contributes to the outcome of hepatitis B virus infection. J. Virol. 2009, 83, 9652–9662. [Google Scholar] [CrossRef] [Green Version]
- Jilbert, A.R.; Miller, D.S.; Scougall, C.A.; Turnbull, H.; Burrell, C.J. Kinetics of duck hepatitis B virus infection following low dose virus inoculation: One virus DNA genome is infectious in neonatal ducks. Virology 1996, 226, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [Green Version]
- Decorsière, A.; Mueller, H.; Van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386. [Google Scholar] [CrossRef]
- Moolla, N.; Kew, M.; Arbuthnot, P. Regulatory elements of hepatitis B virus transcription. J. Viral. Hepat. 2002, 9, 323–331. [Google Scholar] [CrossRef]
- Lamontagne, R.J.; Bagga, S.; Bouchard, M.J. Hepatitis B virus molecular biology and pathogenesis. Hepatoma Res. 2016, 2, 163–186. [Google Scholar] [CrossRef]
- Testoni, B.; Lebosse, F.; Scholtes, C.; Berby, F.; Miaglia, C.; Subic, M.; Loglio, A.; Facchetti, F.; Lampertico, P.; Levrero, M.; et al. Serum hepatitis B core-related antigen (HBcrAg) correlates with covalently closed circular DNA transcriptional activity in chronic hepatitis B patients. J. Hepatol. 2019, 70, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhou, B.; Valdes, J.D.; Sun, J.; Guo, H. Serum Hepatitis B Virus RNA: A New Potential Biomarker for Chronic Hepatitis B Virus Infection. Hepatology 2019, 69, 1816–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Wu, D.; Zhou, L.; Chen, E.; Liu, C.; Tang, X.; Jiang, W.; Han, N.; Li, H.; Tang, H. Present and Future Therapies for Chronic Hepatitis B. Adv. Exp. Med. Biol. 2020, 1179, 137–186. [Google Scholar] [PubMed]
- Ely, A.; Moyo, B.; Arbuthnot, P. Progress With Developing Use of Gene Editing To Cure Chronic Infection With Hepatitis B Virus. Mol. Ther. 2016, 24, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Nijampatnam, B.; Liotta, D.C. Recent advances in the development of HBV capsid assembly modulators. Curr. Opin. Chem. Biol. 2019, 50, 73–79. [Google Scholar] [CrossRef]
- Gripon, P.; Cannie, I.; Urban, S. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J. Virol. 2005, 79, 1613–1622. [Google Scholar] [CrossRef] [Green Version]
- Petersen, J.; Dandri, M.; Mier, W.; Lutgehetmann, M.; Volz, T.; von Weizsacker, F.; Haberkorn, U.; Fischer, L.; Pollok, J.M.; Erbes, B.; et al. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat. Biotechnol. 2008, 26, 335–341. [Google Scholar] [CrossRef]
- Volz, T.; Allweiss, L.; Ben, M.M.; Warlich, M.; Lohse, A.W.; Pollok, J.M.; Alexandrov, A.; Urban, S.; Petersen, J.; Lutgehetmann, M.; et al. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J. Hepatol. 2013, 58, 861–867. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human Argonaute2 Mediates RNA Cleavage Targeted by miRNAs and siRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Zamore, P.D.; Tuschl, T.; Sharp, P.A.; Bartel, D.P. RNAi: Double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 2000, 101, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Elbashir, S.M.; Lendeckel, W.; Tuschl, T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001, 15, 188–200. [Google Scholar] [CrossRef] [Green Version]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Lee, N.S.; Dohjima, T.; Bauer, G.; Li, H.; Li, M.J.; Ehsani, A.; Salvaterra, P.; Rossi, J. Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat. Biotechnol. 2002, 20, 500–505. [Google Scholar] [CrossRef]
- Brummelkamp, T.R.; Bernards, R.; Agami, R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002, 296, 550–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giladi, H.; Ketzinel-Gilad, M.; Rivkin, L.; Felig, Y.; Nussbaum, O.; Galun, E. Small interfering RNA inhibits hepatitis B virus replication in mice. Mol. Ther. 2003, 8, 769–776. [Google Scholar] [CrossRef]
- Hamasaki, K.; Nakao, K.; Matsumoto, K.; Ichikawa, T.; Ishikawa, H.; Eguchi, K. Short interfering RNA-directed inhibition of hepatitis B virus replication. FEBS Lett. 2003, 543, 51–54. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.; Bock, C.T.; Wedemeyer, H.; Wustefeld, T.; Locarnini, S.; Dienes, H.P.; Kubicka, S.; Manns, M.P.; Trautwein, C. Inhibition of hepatitis B virus replication in vivo by nucleoside analogues and siRNA. Gastroenterology 2003, 125, 9–18. [Google Scholar] [CrossRef]
- Konishi, M.; Wu, C.H.; Wu, G.Y. Inhibition of HBV replication by siRNA in a stable HBV-producing cell line. Hepatology 2003, 38, 842–850. [Google Scholar] [CrossRef]
- Qian, Z.K.; Xuan, B.Q.; Min, T.S.; Xu, J.F.; Li, L.; Huang, W.D. Cost-effective method of siRNA preparation and its application to inhibit hepatitis B virus replication in HepG2 cells. World J. Gastroenterol. 2005, 11, 1297–1302. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Martinez, J.; Patkaniowska, A.; Lendeckel, W.; Tuschl, T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 2001, 20, 6877–6888. [Google Scholar] [CrossRef] [Green Version]
- Morrissey, D.V.; Blanchard, K.; Shaw, L.; Jensen, K.; Lockridge, J.A.; Dickinson, B.; McSwiggen, J.A.; Vargeese, C.; Bowman, K.; Shaffer, C.S; et al. Activity of stabilized short interfering RNA in a mouse model of hepatitis B virus replication. Hepatology 2005, 41, 1349–1356. [Google Scholar] [CrossRef]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar] [CrossRef]
- Hean, J.; Crowther, C.; Ely, A.; Ul Islam, R.; Barichievy, S.; Bloom, K.; Weinberg, M.S.; van Otterlo, W.A.; de Koning, C.B.; Salazar, F.; et al. Inhibition of hepatitis B virus replication in vivo using lipoplexes containing altritol-modified antiviral siRNAs. Artif. DNA PNA XNA 2010, 1, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Marimani, M.D.; Ely, A.; Buff, M.C.; Bernhardt, S.; Engels, J.W.; Arbuthnot, P. Inhibition of hepatitis B virus replication in cultured cells and in vivo using 2’-O-guanidinopropyl modified siRNAs. Bioorg. Med. Chem. 2013, 21, 6145–6155. [Google Scholar] [CrossRef] [PubMed]
- Marimani, M.D.; Ely, A.; Buff, M.C.; Bernhardt, S.; Engels, J.W.; Scherman, D.; Escriou, V.; Arbuthnot, P. Inhibition of replication of hepatitis B virus in transgenic mice following administration of hepatotropic lipoplexes containing guanidinopropyl-modified siRNAs. J. Control Release 2015, 209, 198–206. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, A.P.; Nakai, H.; Pandey, K.; Huang, Z.; Salazar, F.H.; Xu, H.; Wieland, S.F.; Marion, P.L.; Kay, M.A. Inhibition of hepatitis B virus in mice by RNA interference. Nat. Biotechnol. 2003, 21, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef]
- Shlomai, A.; Shaul, Y. Inhibition of hepatitis B virus expression and replication by RNA interference. Hepatology 2003, 37, 764–770. [Google Scholar] [CrossRef]
- Dyer, V.; Ely, A.; Bloom, K.; Weinberg, M.; Arbuthnot, P. tRNA Lys3 promoter cassettes that efficiently express RNAi-activating antihepatitis B virus short hairpin RNAs. Biochem. Biophys. Res. Commun. 2010, 398, 640–646. [Google Scholar] [CrossRef]
- Ely, A.; Naidoo, T.; Mufamadi, S.; Crowther, C.; Arbuthnot, P. Expressed anti-HBV primary microRNA shuttles inhibit viral replication efficiently in vitro and in vivo. Mol. Ther. 2008, 16, 1105–1112. [Google Scholar] [CrossRef]
- Ely, A.; Naidoo, T.; Arbuthnot, P. Efficient silencing of gene expression with modular trimeric Pol II expression cassettes comprising microRNA shuttles. Nucleic Acids Res. 2009, 37, e91. [Google Scholar] [CrossRef] [Green Version]
- Carmona, S.; Ely, A.; Crowther, C.; Moolla, N.; Salazar, F.H.; Marion, P.L.; Ferry, N.; Weinberg, M.S.; Arbuthnot, P. Effective inhibition of HBV replication in vivo by anti-HBx short hairpin RNAs. Mol. Ther. 2006, 13, 411–421. [Google Scholar] [CrossRef]
- Uprichard, S.L.; Boyd, B.; Althage, A.; Chisari, F.V. Clearance of hepatitis B virus from the liver of transgenic mice by short hairpin RNAs. Proc. Natl. Acad. Sci. USA 2005, 102, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Ivacik, D.; Ely, A.; Ferry, N.; Arbuthnot, P. Sustained inhibition of hepatitis B virus replication in vivo using RNAi-activating lentiviruses. Gene Ther. 2015, 22, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maepa, M.B.; Ely, A.; Grayson, W.; Arbuthnot, P. Sustained Inhibition of HBV Replication In Vivo after Systemic Injection of AAVs Encoding Artificial Antiviral Primary MicroRNAs. Mol. Ther. Nucleic Acids 2017, 7, 190–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramvis, A. Genotypes and genetic variability of hepatitis B virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, K.; Tanaka, Y.; Kurbanov, F.; Sugauchi, F.; Mano, S.; Maeshiro, T.; Nakayoshi, T.; Wakuta, M.; Miyakawa, Y.; Mizokami, M. A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a Japanese patient and provisionally assigned to new genotype J. J. Virol. 2009, 83, 10538–10547. [Google Scholar] [CrossRef] [Green Version]
- Kimbi, G.C.; Kramvis, A.; Kew, M.C. Distinctive sequence characteristics of subgenotype A1 isolates of hepatitis B virus from South Africa. J. Gen. Virol. 2004, 85, 1211–1220. [Google Scholar] [CrossRef]
- Chan, H.L.; Hui, A.; Wong, M.; Tse, A.M.; Hung, L.C.; Wong, V.W.; Sung, J.J. Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut 2004, 53, 1494–1498. [Google Scholar] [CrossRef] [Green Version]
- Kramvis, A.; Kew, M. Relationship of genotypes of hepatitis B virus to mutations, disease progression and response to antiviral therapy. J. Viral Hepat. 2005, 12, 456–464. [Google Scholar] [CrossRef]
- Erhardt, A.; Blondin, D.; Hauck, K.; Sagir, A.; Kohnle, T.; Heintges, T.; Häussinger, D. Response to interferon alfa is hepatitis B virus genotype dependent: Genotype A is more sensitive to interferon than genotype D. Gut 2005, 54, 1009–1013. [Google Scholar] [CrossRef] [Green Version]
- Cuenca-Gómez, J.Á.; Lozano-Serrano, A.B.; Cabezas-Fernández, M.T.; Soriano-Pérez, M.J.; Vázquez-Villegas, J.; Estévez-Escobar, M.; Cabeza-Barrera, I.; Salas-Coronas, J. Chronic hepatitis B genotype E in African migrants: Response to nucleos (t) ide treatment in real clinical practice. BMC Infect. Dis. 2018, 18, 568. [Google Scholar] [CrossRef] [Green Version]
- Shen, F.; Li, Y.; Wang, Y.; Sozzi, V.; Revill, P.A.; Liu, J.; Gao, L.; Yang, G.; Lu, M.; Sutter, K.; et al. Hepatitis B virus sensitivity to interferon-α in hepatocytes is more associated with cellular interferon response than with viral genotype. Hepatology 2018, 67, 1237–1252. [Google Scholar] [CrossRef] [Green Version]
- Kramvis, A.; Kostaki, E.-G.; Hatzakis, A.; Paraskevis, D. Immunomodulatory function of HBeAg related to short-sighted evolution, transmissibility, and clinical manifestation of hepatitis B virus. Front. Microbiol. 2018, 9, 2521. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Xu, Z.; Liu, Y.; Li, X.; Bai, S.; Ding, N.; Zhong, Y.; Wang, L.; Mao, P.; Zoulim, F.; et al. Hepatitis B virus genotype and basal core promoter/precore mutations are associated with hepatitis B-related acute-on-chronic liver failure without pre-existing liver cirrhosis. J. Viral Hepat. 2010, 17, 887–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Rösler, C.; Kidd-Ljunggren, K.; Nassal, M. Quantitative assessment of the antiviral potencies of 21 shRNA vectors targeting conserved, including structured, hepatitis B virus sites. J. Hepatol. 2010, 52, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Cheng, T.; Cai, Y.-J.; Yuan, Q.; Liu, C.; Zhang, T.; Xia, D.-Z.; Li, R.-Y.; Yang, L.-W.; Wang, Y.-B. RNA Interference inhibits hepatitis B virus of different genotypes in vitro and in vivo. BMC Microbiol. 2010, 10, 214. [Google Scholar] [CrossRef] [Green Version]
- Glebe, D.; Urban, S. Viral and cellular determinants involved in hepadnaviral entry. World J. Gastroenterol. 2007, 13, 22–38. [Google Scholar] [CrossRef] [Green Version]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [Green Version]
- Guillouzo, A.; Corlu, A.; Aninat, C.; Glaise, D.; Morel, F.; Guguen-Guillouzo, C. The human hepatoma HepaRG cells: A highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem. Biol. Interact. 2007, 168, 66–73. [Google Scholar] [CrossRef]
- Sells, M.A.; Chen, M.L.; Acs, G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. USA 1987, 84, 1005–1009. [Google Scholar] [CrossRef] [Green Version]
- Sells, M.A.; Chen, M.L.; Acs, G. Replicative intermediates of hepatitis B virus in HepG2 cells that produce infectious virions. J. Virol. 1988, 62, 2836–2844. [Google Scholar] [CrossRef] [Green Version]
- Hirschman, S.Z.; Price, P.; Garfinkel, E.; Christman, J.; Acs, G. Expression of cloned hepatitis B virus DNA in human cell cultures. Proc. Natl. Acad. Sci. USA 1980, 77, 5507–5511. [Google Scholar] [CrossRef] [Green Version]
- Ladner, S.K.; Otto, M.J.; Barker, C.S.; Zaifert, K.; Wang, G.H.; Guo, J.T.; Seeger, C.; King, R.W. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: A novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 1997, 41, 1715–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huch, M.; Dorrell, C.; Boj, S.F.; van Es, J.H.; Li, V.S.; van de Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J; et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef]
- Liu, F.; Song, Y.; Liu, D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999, 6, 1258–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.L.; Althage, A.; Chung, J.; Chisari, F.V. Hydrodynamic injection of viral DNA: A mouse model of acute hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2002, 99, 13825–13830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Yu, H.; Li, C.; Hirsch, M.L.; Zhang, L.; Samulski, R.J.; Li, W.; Liu, Z. Adeno-associated virus vector mediated delivery of the HBV genome induces chronic hepatitis B virus infection and liver fibrosis in mice. PLoS ONE 2015, 10, e0130052. [Google Scholar] [CrossRef] [Green Version]
- Marion, P.; Salazar, F.; Liittschwager, K.; Bordier, B.; Seeger, C.; Winters, M.; Cooper, A.; Cullen, J. A transgenic mouse lineage useful for testing antivirals targeting hepatitis B virus. In Frontiers in Viral Hepatitis; Elsevier: Amsterdam, The Netherlands, 2003; pp. 197–210. [Google Scholar]
- Guo, W.N.; Zhu, B.; Ai, L.; Yang, D.L.; Wang, B.J. Animal models for the study of hepatitis B virus infection. Zool Res. 2018, 39, 25–31. [Google Scholar]
- Von Weizsäcker, F.; Kock, J.; MacNelly, S.; Ren, S.; Blum, H.E.; Nassal, M. The tupaia model for the study of hepatitis B virus: Direct infection and HBV genome transduction of primary tupaia hepatocytes. Methods Mol. Med. 2004, 96, 153–161. [Google Scholar]
- Wieland, S.F. The chimpanzee model for hepatitis B virus infection. Cold Spring Harb. Perspect Med. 2015, 5, a021469. [Google Scholar] [CrossRef] [Green Version]
- Burwitz, B.J.; Wettengel, J.M.; Muck-Hausl, M.A.; Ringelhan, M.; Ko, C.; Festag, M.M.; Hammond, K.B.; Northrup, M.; Bimber, B.N.; Jacob, T.; et al. Hepatocytic expression of human sodium-taurocholate cotransporting polypeptide enables hepatitis B virus infection of macaques. Nat. Commun. 2017, 8, 2146. [Google Scholar] [CrossRef]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Carmona, S.; Jorgensen, M.R.; Kolli, S.; Crowther, C.; Salazar, F.H.; Marion, P.L.; Fujino, M.; Natori, Y.; Thanou, M.; Arbuthnot, P.; et al. Controlling HBV replication in vivo by intravenous administration of triggered PEGylated siRNA-nanoparticles. Mol. Pharm. 2009, 6, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Rensen, P.C.; van Leeuwen, S.H.; Sliedregt, L.A.; van Berkel, T.J.; Biessen, E.A. Design and synthesis of novel N-acetylgalactosamine-terminated glycolipids for targeting of lipoproteins to the hepatic asialoglycoprotein receptor. J. Med. Chem. 2004, 47, 5798–5808. [Google Scholar] [CrossRef] [PubMed]
- Wooddell, C.I.; Rozema, D.B.; Hossbach, M.; John, M.; Hamilton, H.L.; Chu, Q.; Hegge, J.O.; Klein, J.J.; Wakefield, D.H.; Oropeza, C.E.; et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol. Ther. 2013, 21, 973–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schluep, T.; Lickliter, J.; Hamilton, J.; Lewis, D.L.; Lai, C.L.; Lau, J.Y.; Locarnini, S.A.; Gish, R.G.; Given, B.D. Safety, Tolerability, and Pharmacokinetics of ARC-520 Injection, an RNA Interference-Based Therapeutic for the Treatment of Chronic Hepatitis B Virus Infection, in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2017, 6, 350–362. [Google Scholar] [CrossRef] [Green Version]
- Wooddell, C.I.; Yuen, M.F.; Chan, H.L.; Gish, R.G.; Locarnini, S.A.; Chavez, D.; Ferrari, C.; Given, B.D.; Hamilton, J.; Kanner, S.B.; et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [Green Version]
- Nair, J.K.; Attarwala, H.; Sehgal, A.; Wang, Q.; Aluri, K.; Zhang, X.; Gao, M.; Liu, J.; Indrakanti, R.; Schofield, S.; et al. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates. Nucleic Acids Res. 2017, 45, 10969–10977. [Google Scholar] [CrossRef] [Green Version]
- Foster, D.J.; Brown, C.R.; Shaikh, S.; Trapp, C.; Schlegel, M.K.; Qian, K.; Sehgal, A.; Rajeev, K.G.; Jadhav, V.; Manoharan, M.; et al. Advanced siRNA Designs Further Improve In Vivo Performance of GalNAc-siRNA Conjugates. Mol. Ther. 2018, 26, 708–717. [Google Scholar] [CrossRef]
- Hassler, M.R.; Turanov, A.A.; Alterman, J.F.; Haraszti, R.A.; Coles, A.H.; Osborn, M.F.; Echeverria, D.; Nikan, M.; Salomon, W.E.; Roux, L.; et al. Comparison of partially and fully chemically-modified siRNA in conjugate-mediated delivery in vivo. Nucleic Acids Res. 2018, 46, 2185–2196. [Google Scholar] [CrossRef]
- Yuen, M.F.; Schiefke, I.; Yoon, J.H.; Ahn, S.H.; Heo, J.; Kim, J.H.; Lik Yuen Chan, H.; Yoon, K.T.; Klinker, H.; Manns, M.; et al. RNA Interference Therapy With ARC-520 Results in Prolonged Hepatitis B Surface Antigen Response in Patients With Chronic Hepatitis B Infection. Hepatology 2020, 72, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kootstra, N.A.; Verma, I.M. Gene therapy with viral vectors. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 413–439. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Datta, S.; Imbeaud, S.; Franconi, A.; Mallet, M.; Couchy, G.; Letouze, E.; Pilati, C.; Verret, B.; Blanc, J.F.; et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015, 47, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Logan, G.J.; Dane, A.P.; Hallwirth, C.V.; Smyth, C.M.; Wilkie, E.E.; Amaya, A.K.; Zhu, E.; Khandekar, N.; Ginn, S.L.; Liao, S.H.Y; et al. Identification of liver-specific enhancer-promoter activity in the 3’ untranslated region of the wild-type AAV2 genome. Nat. Genet. 2017, 49, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, J.M., 2nd; Hu, P.; Caballero, S.; Moldovan, L.; Verma, A.; Oudit, G.Y.; Li, Q.; Grant, M.B. Adeno-Associated Virus Overexpression of Angiotensin-Converting Enzyme-2 Reverses Diabetic Retinopathy in Type 1 Diabetes in Mice. Am. J. Pathol. 2016, 186, 1688–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassner, U.; Hollstein, T.; Grenkowitz, T.; Wuhle-Demuth, M.; Salewsky, B.; Demuth, I.; Dippel, M.; Steinhagen-Thiessen, E. Gene Therapy in Lipoprotein Lipase Deficiency: Case Report on the First Patient Treated with Alipogene Tiparvovec Under Daily Practice Conditions. Hum. Gene Ther. 2018, 29, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Castle, M.J.; Turunen, H.T.; Vandenberghe, L.H.; Wolfe, J.H. Controlling AAV Tropism in the Nervous System with Natural and Engineered Capsids. Methods Mol. Biol. 2016, 1382, 133–149. [Google Scholar]
- McCarty, D.M.; Monahan, P.E.; Samulski, R.J. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001, 8, 1248–1254. [Google Scholar] [CrossRef] [Green Version]
- Michler, T.; Grosse, S.; Mockenhaupt, S.; Roder, N.; Stuckler, F.; Knapp, B.; Ko, C.; Heikenwalder, M.; Protzer, U.; Grimm, D. Blocking sense-strand activity improves potency, safety and specificity of anti-hepatitis B virus short hairpin RNA. EMBO Mol. Med. 2016, 8, 1082–1098. [Google Scholar] [CrossRef]
- Mnyandu, N.; Arbuthnot, P.; Maepa, M.B. In vivo delivery of cassettes encoding anti-hbv primary micrornas using an ancestral adeno-associated viral vector. Methods Mol. Biol. 2020, 2115, 171–183. [Google Scholar]
- Hosel, M.; Lucifora, J.; Michler, T.; Holz, G.; Gruffaz, M.; Stahnke, S.; Zoulim, F.; Durantel, D.; Heikenwalder, M.; Nierhoff, D.; et al. Hepatitis B virus infection enhances susceptibility toward adeno-associated viral vector transduction in vitro and in vivo. Hepatology 2014, 59, 2110–2120. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Kan, F.; Yan, T.; Cao, J.; Zhang, L.; Wu, Z.; Li, W. Enhanced antiviral and antifibrotic effects of short hairpin RNAs targeting HBV and TGF-beta in HBV-persistent mice. Sci. Rep. 2017, 7, 3860. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Sun, C.P.; Ma, H.I.; Fang, C.C.; Wu, P.Y.; Xiao, X.; Tao, M.H. Comparative study of anti-hepatitis B virus RNA interference by double-stranded adeno-associated virus serotypes 7, 8, and 9. Mol. Ther. 2009, 17, 352–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mockenhaupt, S.; Grosse, S.; Rupp, D.; Bartenschlager, R.; Grimm, D. Alleviation of off-target effects from vector-encoded shRNAs via codelivered RNA decoys. Proc. Natl. Acad. Sci. USA 2015, 112, E4007–E4016. [Google Scholar] [CrossRef] [Green Version]
- Calcedo, R.; Morizono, H.; Wang, L.; McCarter, R.; He, J.; Jones, D.; Batshaw, M.L.; Wilson, J.M. Adeno-associated virus antibody profiles in newborns, children, and adolescents. Clin. Vaccine Immunol. 2011, 18, 1586–1588. [Google Scholar] [CrossRef] [Green Version]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef]
- Fitzpatrick, Z.; Leborgne, C.; Barbon, E.; Masat, E.; Ronziti, G.; van Wittenberghe, L.; Vignaud, A.; Collaud, F.; Charles, S.; Sola, M.; et al. Influence of Pre-existing Anti-Capsid Neutralizing and Binding Antibodies on AAV Vector Transduction. Mol. Ther. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Lisowski, L.; Dane, A.P.; Chu, K.; Zhang, Y.; Cunningham, S.C.; Wilson, E.M.; Nygaard, S.; Grompe, M.; Alexander, I.E.; Kay, M.A. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature 2014, 506, 382–386. [Google Scholar] [CrossRef] [Green Version]
- Santiago-Ortiz, J.; Ojala, D.S.; Westesson, O.; Weinstein, J.R.; Wong, S.Y.; Steinsapir, A.; Kumar, S.; Holmes, I.; Schaffer, D.V. AAV ancestral reconstruction library enables selection of broadly infectious viral variants. Gene Ther. 2015, 22, 934–946. [Google Scholar] [CrossRef] [Green Version]
- Zinn, E.; Pacouret, S.; Khaychuk, V.; Turunen, H.T.; Carvalho, L.S.; Andres-Mateos, E.; Shah, S.; Shelke, R.; Maurer, A.C.; Plovie, E.; et al. In Silico Reconstruction of the Viral Evolutionary Lineage Yields a Potent Gene Therapy Vector. Cell Rep. 2015, 12, 1056–1068. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wu, S.; Albright, B.; Hirsch, M.; Li, W.; Tseng, Y.S.; Agbandje-McKenna, M.; McPhee, S.; Asokan, A.; Samulski, R.J. Development of Patient-specific AAV Vectors After Neutralizing Antibody Selection for Enhanced Muscle Gene Transfer. Mol. Ther. 2016, 24, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mingozzi, F.; Anguela, X.M.; Pavani, G.; Chen, Y.; Davidson, R.J.; Hui, D.J.; Yazicioglu, M.; Elkouby, L.; Hinderer, C.J.; Faella, A.; et al. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci. Transl. Med. 2013, 5, 194ra92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| siRNA Activator | Delivery | Company | Phase | Identifier (Clinicaltrials.Gov) | End Date | Reference |
|---|---|---|---|---|---|---|
| JNJ-3989 (ARO-HBV) | GalNAc | Arrowhead Pharmaceuticals | I/IIa | NCT03365947 NCT03982186 NCT04129554 | September 2020 | - |
| ARC-520 | GalNAc | Arrowhead Pharmaceuticals | I, IIb | NCT01872065 NCT02065336 NCT02604199 NCT02604212 | Completed/terminated | [96,97,102] |
| ARC-521 | GalNAc | Arrowhead Pharmaceuticals | I | NCT02797522 | Terminated | - |
| VIR-2218 (ALN-HBV02) | GalNAc | Alnylam Pharmaceuticals/ Vir Biotechnology | I/II | NCT03672188 NCT02826018 | March 2021 | - |
| ARB-1467 | LNP | Arbutus Biopharma | IIa | NCT02631096 | Completed | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van den Berg, F.; Limani, S.W.; Mnyandu, N.; Maepa, M.B.; Ely, A.; Arbuthnot, P. Advances with RNAi-Based Therapy for Hepatitis B Virus Infection. Viruses 2020, 12, 851. https://doi.org/10.3390/v12080851
van den Berg F, Limani SW, Mnyandu N, Maepa MB, Ely A, Arbuthnot P. Advances with RNAi-Based Therapy for Hepatitis B Virus Infection. Viruses. 2020; 12(8):851. https://doi.org/10.3390/v12080851
Chicago/Turabian Stylevan den Berg, Fiona, Shonisani Wendy Limani, Njabulo Mnyandu, Mohube Betty Maepa, Abdullah Ely, and Patrick Arbuthnot. 2020. "Advances with RNAi-Based Therapy for Hepatitis B Virus Infection" Viruses 12, no. 8: 851. https://doi.org/10.3390/v12080851
APA Stylevan den Berg, F., Limani, S. W., Mnyandu, N., Maepa, M. B., Ely, A., & Arbuthnot, P. (2020). Advances with RNAi-Based Therapy for Hepatitis B Virus Infection. Viruses, 12(8), 851. https://doi.org/10.3390/v12080851