Lentiviral Vector Pseudotypes: Precious Tools to Improve Gene Modification of Hematopoietic Cells for Research and Gene Therapy




Abstract
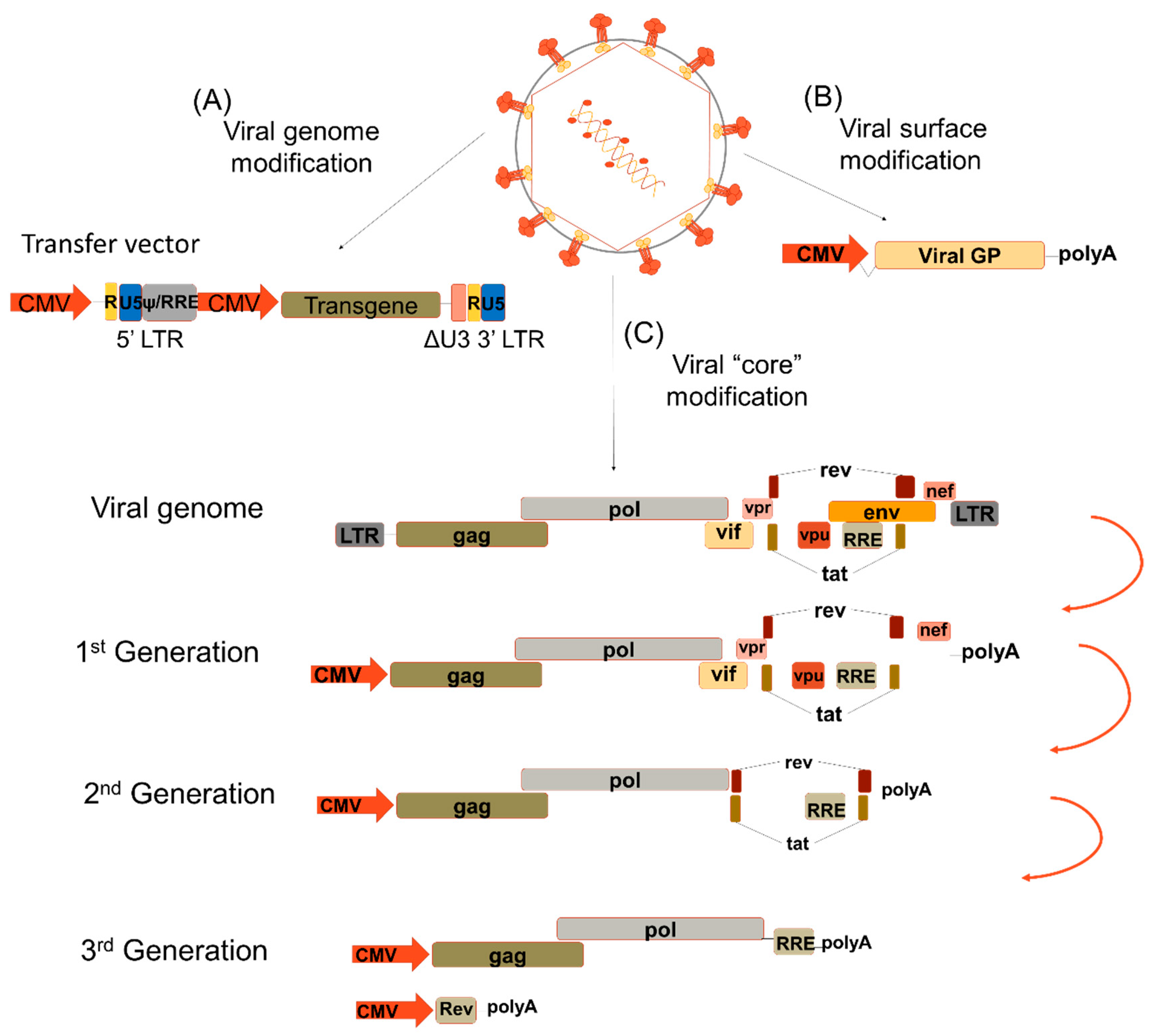
:1. Introduction
2. Pseudotyping
2.1. Pseudotyping of LVs with Baboon Endogenous Virus and Feline Endogenous Virus
2.2. Pseudotyping LVs with H and F Glycoprotein from Measles Virus
2.3. Pseudotyping LVs with Nipah Virus Envelopes
2.4. Cocal Virus
2.5. Envelope Glycoproteins Retargeted to Specific Hematopoietic Cells
3. Gene Therapy
3.1. Introduction to Gene Therapy
3.2. Hematopoietic Stem Cell-Based Gene Therapy
3.3. Gene Therapy Using T Cells
3.3.1. Why Are T Cells Important Target Cells for Gene Therapy?
3.3.2. Novel LV Pseudotypes Allow Efficient T Cell Transduction for Gene Therapy
3.3.3. Chimeric Antigen Receptor T Cells
3.4. B Cells as Gene Therapy Targets
3.5. Gene Therapy Using Natural Killer Cells
4. Gene Editing: A New Upcoming Tool for Gene Therapy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chen, Y.H.; Keiser, M.S.; Davidson, B.L. Viral Vectors for Gene Transfer. Curr. Protoc. Mouse Biol. 2018, 8, e58. [Google Scholar] [CrossRef] [PubMed]
- Verhoeyen, E. Advances in foamy virus vector technology and disease correction could speed the path to clinical application. Mol. Ther. 2012, 20, 1105–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, R.; Mulligan, R.C.; Baltimore, D. Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell 1983, 33, 153–159. [Google Scholar] [CrossRef]
- Escors, D.; Breckpot, K. UKPMC Funders Group Author Manuscript Lentiviral vectors in gene therapy: Their current status and future potential. Arch. Immunol. Ther. Exp. 2011, 58, 107–119. [Google Scholar] [CrossRef] [Green Version]
- High, K.A.; Roncarolo, M.G. Gene therapy. N. Engl. J. Med. 2019, 381, 455–464. [Google Scholar] [CrossRef]
- Schambach, A.; Zychlinski, D.; Ehrnstroem, B.; Baum, C. Biosafety features of lentiviral vectors. Hum. Gene Ther. 2013, 24, 132–142. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef]
- Braun, C.J.; Boztug, K.; Paruzynski, A.; Witzel, M.; Schwarzer, A.; Rothe, M.; Modlich, U.; Beier, R.; Göhring, G.; Steinemann, D.; et al. Gene therapy for Wiskott-Aldrich syndrome--long-term efficacy and genotoxicity. Sci. Transl. Med. 2014, 6, 227ra33. [Google Scholar] [CrossRef]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- Benabdellah, K.; Gutierrez-Guerrero, A.; Cobo, M.; Muñoz, P.; Martín, F. A chimeric HS4-SAR insulator (IS2) that prevents silencing and enhances expression of lentiviral vectors in pluripotent stem cells. PLoS ONE 2014, 9, e84268. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Thrasher, A.J. Gene therapy for PIDs: Progress, pitfalls and prospects. Gene 2013, 525, 174–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Boztug, K.; Schmidt, M.; Schwarzer, A.; Banerjee, P.P.; Díez, I.A.; Dewey, R.A.; Böhm, M.; Nowrouzi, A.; Ball, C.R.; Glimm, H.; et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N. Engl. J. Med. 2010, 363, 1918–1927. [Google Scholar] [CrossRef] [Green Version]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [Green Version]
- Biffi, A.; Bartolomae, C.C.; Cesana, D.; Cartier, N.; Aubourg, P.; Ranzani, M.; Cesani, M.; Benedicenti, F.; Plati, T.; Rubagotti, E.; et al. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood 2011, 117, 5332–5339. [Google Scholar] [CrossRef] [Green Version]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef] [Green Version]
- Río, P.; Navarro, S.; Wang, W.; Sánchez-Domínguez, R.; Pujol, R.M.; Segovia, J.C.; Bogliolo, M.; Merino, E.; Wu, N.; Salgado, R.; et al. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat. Med. 2019, 25, 1396–1401. [Google Scholar] [CrossRef]
- Apolonia, L.; Waddington, S.N.; Fernandes, C.; Ward, N.J.; Bouma, G.; Blundell, M.P.; Thrasher, A.J.; Collins, M.K.; Philpott, N.J. Stable gene transfer to muscle using non-integrating lentiviral vectors. Mol. Ther. 2007, 15, 1947–1954. [Google Scholar] [CrossRef]
- Ortinski, P.I.; O’Donovan, B.; Dong, X.; Kantor, B. Integrase-Deficient Lentiviral Vector as an All-in-One Platform for Highly Efficient CRISPR/Cas9-Mediated Gene Editing. Mol. Ther. Methods Clin. Dev. 2017, 5, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, K.A.; Landau, N.R.; Littman, D.R. Construction and use of a human immunodeficiency virus vector for analysis of virus infectivity. J. Virol. 1990, 64, 5270–5276. [Google Scholar] [CrossRef] [Green Version]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Reiser, J.; Harmison, G.; Kluepfel-Stahl, S.; Brady, R.O.; Karlsson, S.; Schubert, M. Transduction of nondividing cells using pseudotyped defective high-titer HIV type 1 particles. Proc. Natl. Acad. Sci. USA 1996, 93, 15266–15271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkina, R.K.; Walton, R.M.; Chen, M.L.; Li, Q.X.; Planelles, V.; Chen, I.S. High-efficiency gene transfer into CD34+ cells with a human immunodeficiency virus type 1-based retroviral vector pseudotyped with vesicular stomatitis virus envelope glycoprotein G. J. Virol. 1996, 70, 2581–2585. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, R.; Tralka, T.S.; Willingham, M.C.; Pastan, I. Inhibition of VSV binding and infectivity by phosphatidylserine: Is phosphatidylserine a VSV-binding site? Cell 1983, 32, 639–646. [Google Scholar] [CrossRef]
- Coil, D.A.; Miller, A.D. Phosphatidylserine is not the cell surface receptor for vesicular stomatitis virus. J. Virol. 2004, 78, 10920–10926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, J.C.; Friedmann, T.; Driever, W.; Burrascano, M.; Yee, J.K. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: Concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. USA 1993, 90, 8033–8037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhoeyen, E.; Cosset, F.L. Surface-engineering of lentiviral vectors. J. Gene Med. 2004, 6. [Google Scholar] [CrossRef]
- Ozog, S.; Chen, C.X.; Simpson, E.; Garijo, O.; Timberlake, N.D.; Minder, P.; Verhoeyen, E.; Torbett, B.E. CD46 Null Packaging Cell Line Improves Measles Lentiviral Vector Production and Gene Delivery to Hematopoietic Stem and Progenitor Cells. Mol. Ther. Methods Clin. Dev. 2019, 13, 27–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, O.; Gisch, D.W.; Wohlfahrt, M.E.; Adams, A.B.; Greenberg, P.D.; Schmitt, T.M.; Trobridge, G.D.; Kiem, H.P. Development of third-generation cocal envelope producer cell lines for robust lentiviral gene transfer into hematopoietic stem cells and t-cells. Mol. Ther. 2016, 24, 1237–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trobridge, G.D.; Wu, R.A.; Hansen, M.; Ironside, C.; Watts, K.L.; Olsen, P.; Beard, B.C.; Kiem, H.P. Cocal-pseudotyped lentiviral vectors resist inactivation by human serum and efficiently transduce primate hematopoietic repopulating cells. Mol. Ther. 2010, 18, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, I.; Luyet, P.-P.; Pons, V.; Ferguson, C.; Emans, N.; Petiot, A.; Mayran, N.; Demaurex, N.; Fauré, J.; Sadoul, R.; et al. Endosome-to-cytosol transport of viral nucleocapsids. Nat. Cell Biol. 2005, 7, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.S.; Jenni, S.; Stanifer, M.L.; Roth, E.; Whelan, S.P.J.; van Oijen, A.M.; Harrison, S.C. Mechanism of membrane fusion induced by vesicular stomatitis virus G protein. Proc. Natl. Acad. Sci. USA 2017, 114, E28–E36. [Google Scholar] [CrossRef] [Green Version]
- Girard-Gagnepain, A.; Amirache, F.; Costa, C.; Lévy, C.; Frecha, C.; Fusil, F.; Nègre, D.; Lavillette, D.; Cosset, F.L.; Verhoeyen, E. Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood 2014, 124, 1221–1231. [Google Scholar] [CrossRef] [Green Version]
- Munis, A.M.; Tijani, M.; Hassall, M.; Mattiuzzo, G.; Collins, M.K.; Takeuchi, Y. Characterization of Antibody Interactions with the G Protein of Vesicular Stomatitis Virus Indiana Strain and Other Vesiculovirus G Proteins. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Tijani, M.; Munis, A.M.; Perry, C.; Sanber, K.; Ferraresso, M.; Mukhopadhyay, T.; Themis, M.; Nisoli, I.; Mattiuzzo, G.; Collins, M.K.; et al. Lentivector Producer Cell Lines with Stably Expressed Vesiculovirus Envelopes. Mol. Ther. Methods Clin. Dev. 2018, 10, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Radek, C.; Bernadin, O.; Drechsel, K.; Cordes, N.; Pfeifer, R.; Sträßer, P.; Mormin, M.; Gutierrez-Guerrero, A.; Cosset, F.-L.; Kaiser, A.D.; et al. Vectofusin-1 Improves Transduction of Primary Human Cells with Diverse Retroviral and Lentiviral Pseudotypes, Enabling Robust, Automated Closed-System Manufacturing. Hum. Gene Ther. 2019, 30, 1477–1493. [Google Scholar] [CrossRef]
- Colamartino, A.B.L.; Lemieux, W.; Bifsha, P.; Nicoletti, S.; Chakravarti, N.; Sanz, J.; Roméro, H.; Selleri, S.; Béland, K.; Guiot, M.; et al. Efficient and Robust NK-Cell Transduction With Baboon Envelope Pseudotyped Lentivector. Front. Immunol. 2019, 10, 2873. [Google Scholar] [CrossRef]
- Bari, R.; Granzin, M.; Tsang, K.S.; Roy, A.; Krueger, W.; Orentas, R.; Pfeifer, R.; Moeker, N.; Verhoeyen, E.; Dropulic, B.; et al. A distinct subset of highly proliferative and lentiviral vector (LV)-transducible NK cells define a readily engineered subset for adoptive cellular therapy. Front. Immunol. 2019, 10, 2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lévy, C.; Frecha, C.; Costa, C.; Rachinel, N.; Salles, G.; Cosset, F.-L.; Verhoeyen, E. Lentiviral vectors and transduction of human cancer B cells. Blood 2010, 116, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Bernadin, O.; Amirache, F.; Girard-Gagnepain, A.; Moirangthem, R.D.; Lévy, C.; Ma, K.; Costa, C.; Nègre, D.; Reimann, C.; Fenard, D.; et al. Baboon envelope LVs efficiently transduced human adult, fetal, and progenitor T cells and corrected SCID-X1 T-cell deficiency. Blood Adv. 2019, 3, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.; Fusil, F.; Amirache, F.; Costa, C.; Girard-Gagnepain, A.; Negre, D.; Bernadin, O.; Garaulet, G.; Rodriguez, A.; Nair, N.; et al. Baboon envelope pseudotyped lentiviral vectors efficiently transduce human B cells and allow active factor IX B cell secretion in vivo in NOD/SCIDγc-/- mice. J. Thromb. Haemost. 2016, 14, 2478–2492. [Google Scholar] [CrossRef] [Green Version]
- Verhoeyen, E.; Cosset, F.L. Engineering the surface glycoproteins of lentiviral vectors for targeted gene transfer. Cold Spring Harb. Protoc. 2009, 4. [Google Scholar] [CrossRef]
- Frecha, C.; Costa, C.; Nègre, D.; Gauthier, E.; Russell, S.J.; Cosset, F.-L.; Verhoeyen, E. Stable transduction of quiescent T cells without induction of cycle progression by a novel lentiviral vector pseudotyped with measles virus glycoproteins. Blood 2008, 112, 4843–4852. [Google Scholar] [CrossRef] [Green Version]
- Lévy, C.; Amirache, F.; Girard-Gagnepain, A.; Frecha, C.; Roman-Rodríguez, F.J.; Bernadin, O.; Costa, C.; Nègre, D.; Gutierrez-Guerrero, A.; Vranckx, L.S.; et al. Measles virus envelope pseudotyped lentiviral vectors transduce quiescent human HSCs at an efficiency without precedent. Blood Adv. 2017, 1, 2088–2104. [Google Scholar] [CrossRef]
- Zhou, Q.; Uhlig, K.M.; Muth, A.; Kimpel, J.; Lévy, C.; Münch, R.C.; Seifried, J.; Pfeiffer, A.; Trkola, A.; Coulibaly, C.; et al. Exclusive Transduction of Human CD4+ T Cells upon Systemic Delivery of CD4-Targeted Lentiviral Vectors. J. Immunol. 2015, 195, 2493–2501. [Google Scholar] [CrossRef] [Green Version]
- Witting, S.R.; Vallanda, P.; Gamble, A.L. Characterization of a third generation lentiviral vector pseudotyped with Nipah virus envelope proteins for endothelial cell transduction. Gene Ther. 2013, 20, 997–1005. [Google Scholar] [CrossRef] [Green Version]
- Palomares, K.; Vigant, F.; Van Handel, B.; Pernet, O.; Chikere, K.; Hong, P.; Sherman, S.P.; Patterson, M.; An, D.S.; Lowry, W.E.; et al. Nipah Virus Envelope-Pseudotyped Lentiviruses Efficiently Target ephrinB2-Positive Stem Cell Populations In Vitro and Bypass the Liver Sink When Administered In Vivo. J. Virol. 2013, 87, 2094–2108. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, R.; Miyaho, R.N.; Hashimoto, A.; Abe, M.; Yasuda, J.; Miyazawa, T. Suppression of production of baboon endogenous virus by dominant negative mutants of cellular factors involved in multivesicular body sorting pathway. Virus Res. 2015, 196, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Ikeda, Y.; Yonemitsu, Y.; Miyazaki, M.; Inoue, M.; Hasegawa, M.; Sueishi, K.; Ishibashi, T. Inhibition of choroidal neovascularization via brief subretinal exposure to a newly developed lentiviral vector pseudotyped with sendai viral envelope proteins. Hum. Gene Ther. 2010, 21, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Frecha, C.; Costa, C.; Nègre, D.; Amirache, F.; Trono, D.; Rio, P.; Bueren, J.; Cosset, F.L.; Verhoeyen, E. A novel lentiviral vector targets gene transfer into human hematopoietic stem cells in marrow from patients with bone marrow failure syndrome and in vivo in humanized mice. Blood 2012, 119, 1139–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lévy, C.; Amirache, F.; Costa, C.; Frecha, C.; Muller, C.P.; Kweder, H.; Buckland, R.; Cosset, F.L.; Verhoeyen, E. Lentiviral vectors displaying modified measles virus gp overcome pre-existing immunity in in vivo-like transduction of human T and B cells. Mol. Ther. 2012, 20, 1699–1712. [Google Scholar] [CrossRef] [Green Version]
- Amirache, F.; Lévy, C.; Costa, C.; Mangeot, P.E.; Torbett, B.E.; Wang, C.X.; Nègre, D.; Cosset, F.-L.; Verhoeyen, E. Mystery solved: VSV-G-LVs do not allow efficient gene transfer into unstimulated T cells, B. Blood 2014, 123, 1422–1424. [Google Scholar] [CrossRef]
- Kweder, H.; Ainouze, M.; Cosby, S.L.; Muller, C.P.; Lévy, C.; Verhoeyen, E.; Cosset, F.L.; Manet, E.; Buckland, R. Mutations in the H, F, or M proteins can facilitate resistance of measles virus to neutralizing human anti-MV sera. Adv. Virol. 2014, 2014, 205617. [Google Scholar] [CrossRef] [Green Version]
- Frecha, C.; Levy, C.; Costa, C.; Negre, D.; Amirache, F.; Buckland, R.; Russell, S.J.; Cosset, F.-L.; Verhoeyen, E. Measles Virus Glycoprotein-Pseudotyped Lentiviral Vector-Mediated Gene Transfer into Quiescent Lymphocytes Requires Binding to both SLAM and CD46 Entry Receptors. J. Virol. 2011, 85, 5975–5985. [Google Scholar] [CrossRef] [Green Version]
- Frecha, C.; Costa, C.; Lévy, C.; Nègre, D.; Russell, S.J.; Maisner, A.; Salles, G.; Peng, K.-W.; Cosset, F.-L.; Verhoeyen, E. Efficient and stable transduction of resting B lymphocytes and primary chronic lymphocyte leukemia cells using measles virus gp displaying lentiviral vectors. Blood 2009, 114, 3173–3180. [Google Scholar] [CrossRef]
- Labenski, V.; Suerth, J.D.; Barczak, E.; Heckl, D.; Levy, C.; Bernadin, O.; Charpentier, E.; Williams, D.A.; Fehse, B.; Verhoeyen, E.; et al. Alpharetroviral self-inactivating vectors produced by a superinfection-resistant stable packaging cell line allow genetic modification of primary human T lymphocytes. Biomaterials 2016, 97, 97–109. [Google Scholar] [CrossRef]
- Kalodimou, G.; Veit, S.; Jany, S.; Kalinke, U.; Broder, C.C.; Sutter, G.; Volz, A. A soluble version of nipah virus glycoprotein G delivered by vaccinia virus MVA activates specific CD8 and CD4 T cells in mice. Viruses 2019, 12, 26. [Google Scholar] [CrossRef] [Green Version]
- Bender, R.R.; Muth, A.; Schneider, I.C.; Friedel, T.; Hartmann, J.; Plückthun, A.; Maisner, A.; Buchholz, C.J. Receptor-Targeted Nipah Virus Glycoproteins Improve Cell-Type Selective Gene Delivery and Reveal a Preference for Membrane-Proximal Cell Attachment. PLoS Pathog. 2016, 12, e1005641. [Google Scholar] [CrossRef] [PubMed]
- Khetawat, D.; Broder, C.C. A functional henipavirus envelope glycoprotein pseudotyped lentivirus assay system. Virol. J. 2010, 7, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, N.B.; Dimos, J.T.; Schaniel, C.; Hackney, J.A.; Moore, K.A.; Lemischka, I.R. A stem cell molecular signature. Science 2002, 298, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Pauszek, S.J.; Allende, R.; Rodriguez, L.L. Characterization of the full-length genomic sequences of vesicular stomatitis Cocal and Alagoas viruses. Arch. Virol. 2008, 153, 1353–1357. [Google Scholar] [CrossRef] [PubMed]
- Munis, A.M.; Mattiuzzo, G.; Bentley, E.M.; Collins, M.K.; Eyles, J.E.; Takeuchi, Y. Use of Heterologous Vesiculovirus G Proteins Circumvents the Humoral Anti-envelope Immunity in Lentivector-Based In Vivo Gene Delivery. Mol. Ther.-Nucleic Acids 2019, 17, 126–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasaraneni, N.; Chamoun-Emanuelli, A.M.; Wright, G.A.; Chen, Z. A simple strategy for retargeting lentiviral vectors to desired cell types via a disulfide-bond-forming protein-peptide pair. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, K.; Hanauer, J.R.H.; Prüfer, S.; Münch, R.C.; Völker, I.; Filippis, C.; Jost, C.; Hanschmann, K.M.; Cattaneo, R.; Peng, K.W.; et al. DARPin-targeting of measles virus: Unique bispecificity, effective oncolysis, and enhanced safety. Mol. Ther. 2013, 21, 849–859. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Weidner, T.; Thalheimer, F.B.; Buchholz, C.J. In vivo generated human CAR T cells eradicate tumor cells. Oncoimmunology 2019, 8, e1671761. [Google Scholar] [CrossRef] [Green Version]
- Morizono, K.; Xie, Y.; Ringpis, G.-E.; Johnson, M.; Nassanian, H.; Lee, B.; Wu, L.; Chen, I.S.Y. Lentiviral vector retargeting to P-glycoprotein on metastatic melanoma through intravenous injection. Nat. Med. 2005, 11, 346–352. [Google Scholar] [CrossRef]
- Münch, R.C.; Mühlebach, M.D.; Schaser, T.; Kneissl, S.; Jost, C.; Plückthun, A.; Cichutek, K.; Buchholz, C.J. DARPins: An efficient targeting domain for lentiviral vectors. Mol. Ther. 2011, 19, 686–693. [Google Scholar] [CrossRef]
- Zhou, Q.; Schneider, I.C.; Edes, I.; Honegger, A.; Bach, P.; Schönfeld, K.; Schambach, A.; Wels, W.S.; Kneissl, S.; Uckert, W.; et al. T-cell receptor gene transfer exclusively to human CD8+ cells enhances tumor cell killing. Blood 2012, 120, 4334–4342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeiffer, A.; Thalheimer, F.B.; Hartmann, S.; Frank, A.M.; Bender, R.R.; Danisch, S.; Costa, C.; Wels, W.S.; Modlich, U.; Stripecke, R.; et al. In vivo generation of human CD 19-CAR T cells results in B-cell depletion and signs of cytokine release syndrome. EMBO Mol. Med. 2018, 10, e9158. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Gutierrez-Guerrero, A.; Benabdellah, K. No TitleGene Therapy for Primary Immunodeficiencies, Gene Therapy-Tools and Potential Applications; Martin, F., Ed.; IntechOpen: London, UK, 2013; ISBN 978-953-51-1014-9. [Google Scholar]
- Naldini, L. Gene therapy returns to centre stage. Nature 2015, 526, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Weinberg, K.I.; Nolta, J.A.; Heiss, L.N.; Lenarsky, C.; Crooks, G.M.; Hanley, M.E.; Annett, G.; Brooks, J.S.; el-Khoureiy, A. Engraftment of gene-modified umbilical cord blood cells in neonates with adenosine deaminase deficiency. Nat. Med. 1995, 1, 1017–1023. [Google Scholar] [CrossRef] [Green Version]
- Cavazzana, M.; Bushman, F.D.; Miccio, A.; André-Schmutz, I.; Six, E. Gene therapy targeting haematopoietic stem cells for inherited diseases: Progress and challenges. Nat. Rev. Drug Discov. 2019, 18, 447–462. [Google Scholar] [CrossRef] [Green Version]
- Ikawa, Y.; Miccio, A.; Magrin, E.; Kwiatkowski, J.L.; Rivella, S.; Cavazzana, M. Gene therapy of hemoglobinopathies: Progress and future challenges. Concr. J. 1969, 7, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Tang, R.; Xu, Z. Gene therapy: A double-edged sword with great powers. Mol. Cell. Biochem. 2020. [Google Scholar] [CrossRef]
- Kohn, D.B.; Hershfield, M.S.; Puck, J.M.; Aiuti, A.; Blincoe, A.; Gaspar, H.B.; Notarangelo, L.D.; Grunebaum, E. Consensus approach for the management of severe combined immune deficiency caused by adenosine deaminase deficiency. J. Allergy Clin. Immunol. 2019, 143, 852–863. [Google Scholar] [CrossRef]
- Gaspar, H.B.; Cooray, S.; Gilmour, K.C.; Parsley, K.L.; Adams, S.; Howe, S.J.; Al Ghonaium, A.; Bayford, J.; Brown, L.; Davies, E.G.; et al. Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 2011, 3, 97ra79. [Google Scholar] [CrossRef]
- Hacein-Bey Abina, S.; Gaspar, H.B.; Blondeau, J.; Caccavelli, L.; Charrier, S.; Buckland, K.; Picard, C.; Six, E.; Himoudi, N.; Gilmour, K.; et al. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA 2015, 313, 1550–1563. [Google Scholar] [CrossRef]
- Grez, M.; Reichenbach, J.; Schwäble, J.; Seger, R.; Dinauer, M.C.; Thrasher, A.J. Gene therapy of chronic granulomatous disease: The engraftment dilemma. Mol. Ther. 2011, 19, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Konno, A.; Wada, T.; Schurman, S.H.; Garabedian, E.K.; Kirby, M.; Anderson, S.M.; Candotti, F. Differential contribution of Wiskott-Aldrich syndrome protein to selective advantage in T- and B-cell lineages. Blood 2004, 103, 676–678. [Google Scholar] [CrossRef] [PubMed]
- Tolar, J.; Adair, J.E.; Antoniou, M.; Bartholomae, C.C.; Becker, P.S.; Blazar, B.R.; Bueren, J.; Carroll, T.; Cavazzana-Calvo, M.; Clapp, D.W.; et al. Stem cell gene therapy for fanconi anemia: Report from the 1st international fanconi anemia gene therapy working group meeting. Mol. Ther. 2011, 19, 1193–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhoeyen, E.; Roman-Rodriguez, F.J.; Cosset, F.-L.; Levy, C.; Rio, P. Gene Therapy in Fanconi Anemia: A Matter of Time, Safety and Gene Transfer Tool Efficiency. Curr. Gene Ther. 2017, 16, 297–308. [Google Scholar] [CrossRef]
- Tolar, J.; Becker, P.S.; Clapp, D.W.; Hanenberg, H.; De Heredia, C.D.; Kiem, H.P.; Navarro, S.; Qasba, P.; Rio, P.; Schmidt, M.; et al. Gene therapy for fanconi anemia: One step closer to the clinic. Hum. Gene Ther. 2012, 23, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Giardine, B.; Borg, J.; Viennas, E.; Pavlidis, C.; Moradkhani, K.; Joly, P.; Bartsakoulia, M.; Riemer, C.; Miller, W.; Tzimas, G.; et al. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2014, 42, D1063–D1069. [Google Scholar] [CrossRef]
- Drakopoulou, E.; Georgomanoli, M.; Lederer, C.W.; Kleanthous, M.; Costa, C.; Bernadin, O.; Cosset, F.L.; Voskaridou, E.; Verhoeyen, E.; Papanikolaou, E.; et al. A Novel BaEVRless-Pseudotyped γ-Globin Lentiviral Vector Drives High and Stable Fetal Hemoglobin Expression and Improves Thalassemic Erythropoiesis In Vitro. Hum. Gene Ther. 2019, 30, 601–617. [Google Scholar] [CrossRef]
- Frecha, C.; Lévy, C.; Cosset, F.L.; Verhoeyen, E. Advances in the field of lentivector-based transduction of T and B lymphocytes for gene therapy. Mol. Ther. 2010, 18, 1748–1757. [Google Scholar] [CrossRef]
- Maurice, M.; Verhoeyen, E.; Salmon, P.; Trono, D.; Russell, S.J.; Cosset, F.L. Efficient gene transfer into human primary blood lymphocytes by surface-engineered lentiviral vectors that display a T cell-activating polypeptide. Blood 2002, 99, 2342–2350. [Google Scholar] [CrossRef] [Green Version]
- Buchschacher, G.L.J.; Wong-Staal, F. Approaches to gene therapy for human immunodeficiency virus infection. Hum. Gene Ther. 2001, 12, 1013–1019. [Google Scholar] [CrossRef]
- Mhaidly, R.; Verhoeyen, E. The Future: In Vivo CAR T Cell Gene Therapy. Mol. Ther. 2019, 27, 707–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef] [PubMed]
- Jamali, A.; Kapitza, L.; Schaser, T.; Johnston, I.C.D.; Buchholz, C.J.; Hartmann, J. Highly Efficient and Selective CAR-Gene Transfer Using CD4- and CD8-Targeted Lentiviral Vectors. Mol. Ther. Methods Clin. Dev. 2019, 13, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Mhaidly, R.; Verhoeyen, E. Humanized Mice Are Precious Tools for Preclinical Evaluation of CAR T and CAR NK Cell Therapies. Cancers 2020, 12, 1915. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.D.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef] [Green Version]
- Fusil, F.; Calattini, S.; Amirache, F.; Mancip, J.; Costa, C.; Robbins, J.B.; Douam, F.; Lavillette, D.; Law, M.; Defrance, T.; et al. A lentiviral vector allowing physiologically regulated membrane-anchored and secreted antibody expression depending on B-cell maturation status. Mol. Ther. 2015, 23, 1734–1747. [Google Scholar] [CrossRef]
- Venstrom, J.M.; Pittari, G.; Gooley, T.A.; Chewning, J.H.; Spellman, S.; Haagenson, M.; Gallagher, M.M.; Malkki, M.; Petersdorf, E.; Dupont, B.; et al. HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N. Engl. J. Med. 2012, 367, 805–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [Green Version]
- Herberman, R.B.; Nunn, M.E.; Holden, H.T.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int. J. Cancer 1975, 16, 230–239. [Google Scholar] [CrossRef]
- Sutlu, T.; Nyström, S.; Gilljam, M.; Stellan, B.; Applequist, S.E.; Alici, E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: Implications for gene therapy. Hum. Gene Ther. 2012, 23, 1090–1100. [Google Scholar] [CrossRef] [Green Version]
- Afzal, S.; Sirohi, P.; Singh, N.K. A review of CRISPR associated genome engineering: Application, advances and future prospects of genome targeting tool for crop improvement. Biotechnol. Lett. 2020, 42, 1611–1632. [Google Scholar] [CrossRef] [PubMed]
- Vasileva, A.; Jessberger, R. Precise hit: Adeno-associated virus in gene targeting. Nat. Rev. Microbiol. 2005, 3, 837–847. [Google Scholar] [CrossRef] [PubMed]
- van Haasteren, J.; Li, J.; Scheideler, O.J.; Murthy, N.; Schaffer, D.V. The delivery challenge: Fulfilling the promise of therapeutic genome editing. Nat. Biotechnol. 2020, 38, 845–855. [Google Scholar] [CrossRef]
- Gasiunas, G.; Siksnys, V. RNA-dependent DNA endonuclease Cas9 of the CRISPR system: Holy Grail of genome editing? Trends Microbiol. 2013, 21, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.T.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, P.K.; Ferreira, L.M.R.; Collins, R.; Meissner, T.B.; Boutwell, C.L.; Friesen, M.; Vrbanac, V.; Garrison, B.S.; Stortchevoi, A.; Bryder, D.; et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 2014, 15, 643–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, P.; Schiroli, G.; Escobar, G.; Di Tomaso, T.; Firrito, C.; Calabria, A.; Moi, D.; Mazzieri, R.; Bonini, C.; Holmes, M.C.; et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature 2014, 510, 235–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Mangeot, P.E.; Risson, V.; Fusil, F.; Marnef, A.; Laurent, E.; Blin, J.; Mournetas, V.; Massouridès, E.; Sohier, T.J.M.; Corbin, A.; et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 2019, 10, 45. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Guerrero, A.; Mangeot, P.E.; Costa, C.; Bernadin, O.; Froment, G.; Martin, F.; Ricci, E.P.; Cosset, F.-L.; Verhoeyen, E. Efficient Genome Editing in Primary Human T, B and HSCs Using Baboon Envelope Gp Pseudotyped Viral Derived “Nanoblades” Loaded with Cas9/sgRNA Ribonucleoprotein-ASGCT 21st Annual Meeting Abstracts. Mol. Ther. 2018, 26, A116. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Pseudotypes | Original Virus | Receptor | Cell Tropism | Efficiency | References |
|---|---|---|---|---|---|
| VSV-G | Vesicular stomatitis virus | LDL-R | Broad in non-primary cells | High | [29] |
| BaEV | Baboon endogenous retrovirus | ASCT-1 ASCT-2 | CD34+ cells | 30% | [36] |
| Naïve T cells | Up to 80% | [39,40,41] | |||
| Naïve B cells | 40% | [42] | |||
| Memory B cells | 20% | [42] | |||
| Natural killer | 40% | [39,40,41] | |||
| Early thymocytes | Up to 80% | [39,40,41] | |||
| RD114 | Feline endogenous retrovirus | ASCT-2 | Naïve T cells | Up to 60% | [43] |
| Naïve B cells | Up to 30% | [44] | |||
| H/F | Measles virus | SLAM CD46 | CD34+ cells | [30,45] | |
| Resting memory T cells | [46] | ||||
| Naïve T cells | Up to 50% | [46] | |||
| Quiescent B cells | [42,46] | ||||
| Resting HSCs | Up to 70% | [47] | |||
| Dendritic cells | [47,48] | ||||
| G/F | Nipah virus | EphinB2/B3 | Pericytes | 20–40% | [49,50] |
| Tumor endothelium | [49,50] | ||||
| COCV | Cocal virus | LDL-R | Stimulated CD34+ cells | Up to 80% | [32] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutierrez-Guerrero, A.; Cosset, F.-L.; Verhoeyen, E. Lentiviral Vector Pseudotypes: Precious Tools to Improve Gene Modification of Hematopoietic Cells for Research and Gene Therapy. Viruses 2020, 12, 1016. https://doi.org/10.3390/v12091016
Gutierrez-Guerrero A, Cosset F-L, Verhoeyen E. Lentiviral Vector Pseudotypes: Precious Tools to Improve Gene Modification of Hematopoietic Cells for Research and Gene Therapy. Viruses. 2020; 12(9):1016. https://doi.org/10.3390/v12091016
Chicago/Turabian StyleGutierrez-Guerrero, Alejandra, François-Loïc Cosset, and Els Verhoeyen. 2020. "Lentiviral Vector Pseudotypes: Precious Tools to Improve Gene Modification of Hematopoietic Cells for Research and Gene Therapy" Viruses 12, no. 9: 1016. https://doi.org/10.3390/v12091016
APA StyleGutierrez-Guerrero, A., Cosset, F. -L., & Verhoeyen, E. (2020). Lentiviral Vector Pseudotypes: Precious Tools to Improve Gene Modification of Hematopoietic Cells for Research and Gene Therapy. Viruses, 12(9), 1016. https://doi.org/10.3390/v12091016