Discoveries of Exoribonuclease-Resistant Structures of Insect-Specific Flaviviruses Isolated in Zambia

 ,
,  , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site and Sample Collection
2.2. Flavivirus Genome Screening
2.3. Virus Isolation
2.4. Next Generation Sequencing
2.5. Rapid Amplification cDNA Ends Analysis
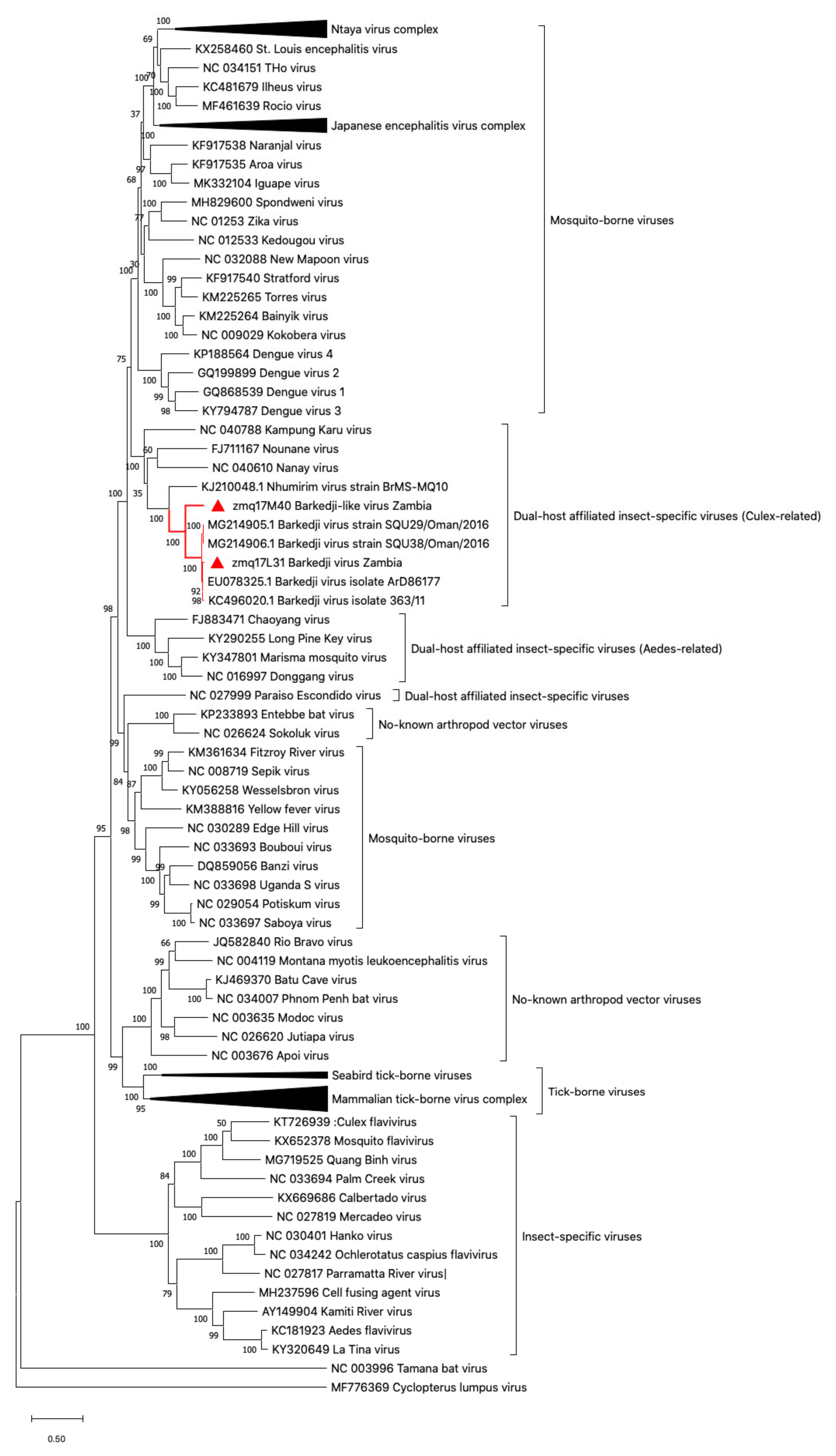
2.6. Phylogenetic Tree Analysis
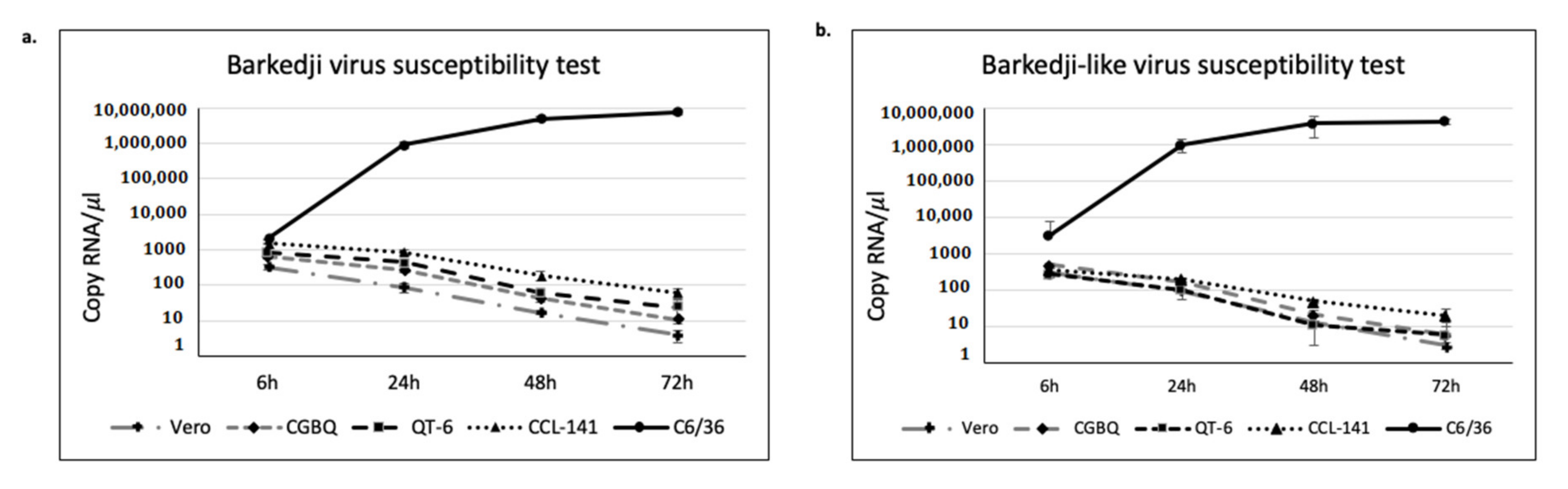
2.7. Virus Susceptibility Test
2.8. Flavivirus Genome Data Set
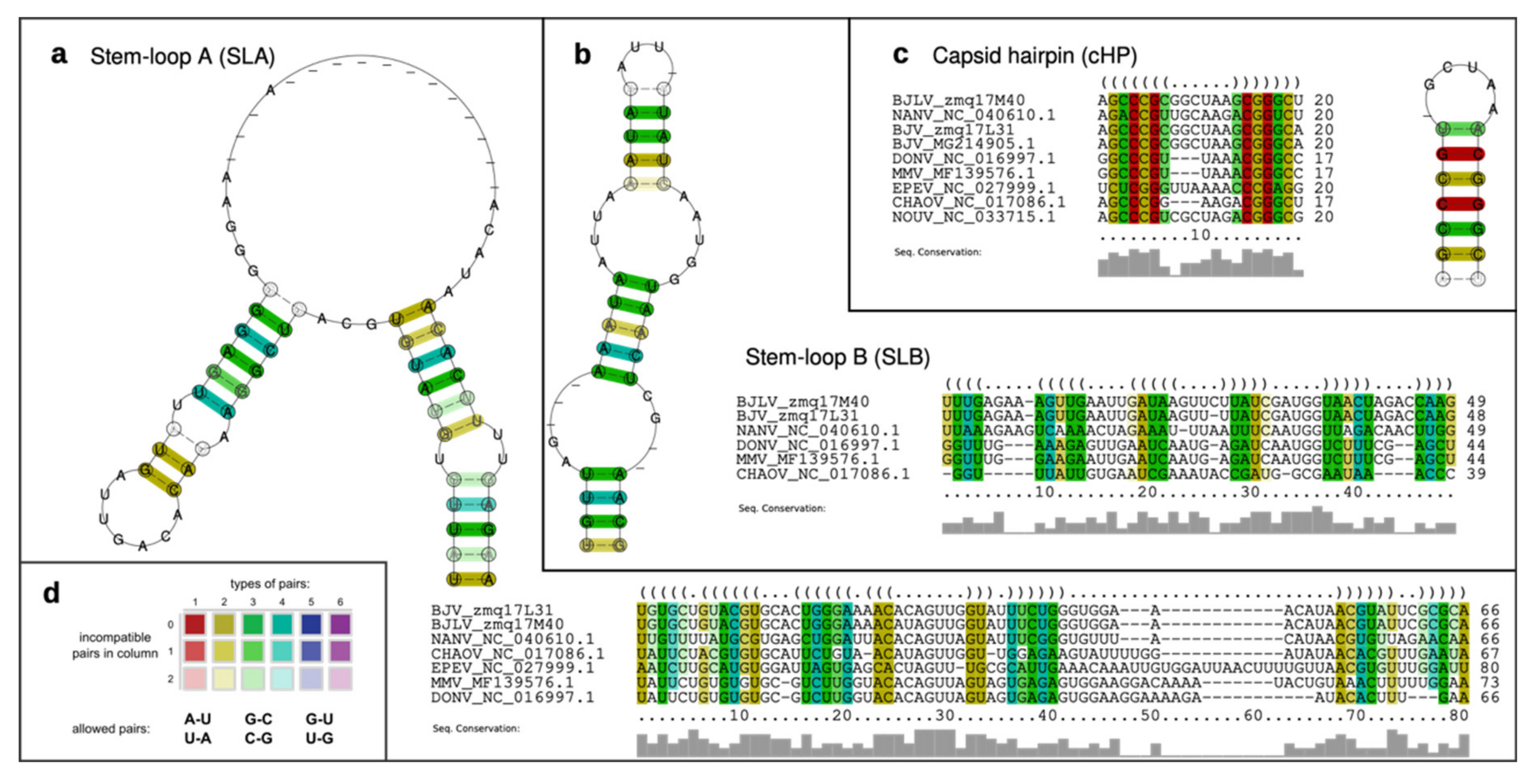
2.9. Characterization of Structurally Homologous RNAs
2.10. In Vitro Xrn1 Resistance Assay
2.11. Xrn1 Stop Point Identification
3. Results
3.1. Flavivirus Genome Detection and Virus Isolation
3.2. Barkedji-Like Virus (BJLV)
3.3. Barkedji Virus (BJV)
3.4. BJV Zambia and BJLV Growth in Mosquito and Vertebrate Cell Lines
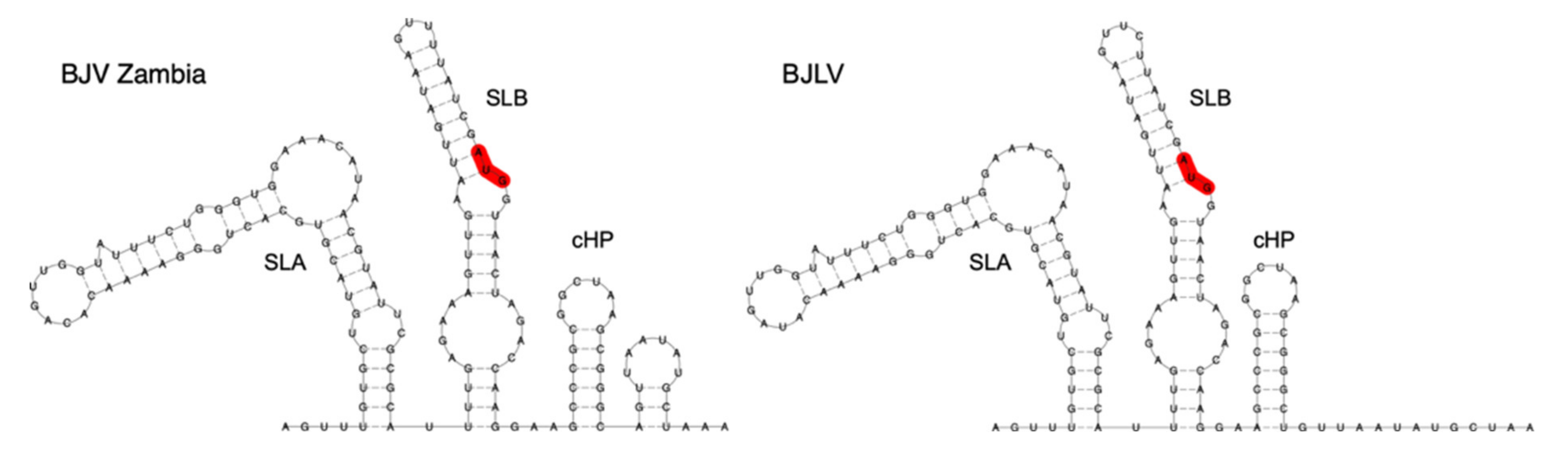
3.5. Secondary Structure Analysis of 5′-UTRs
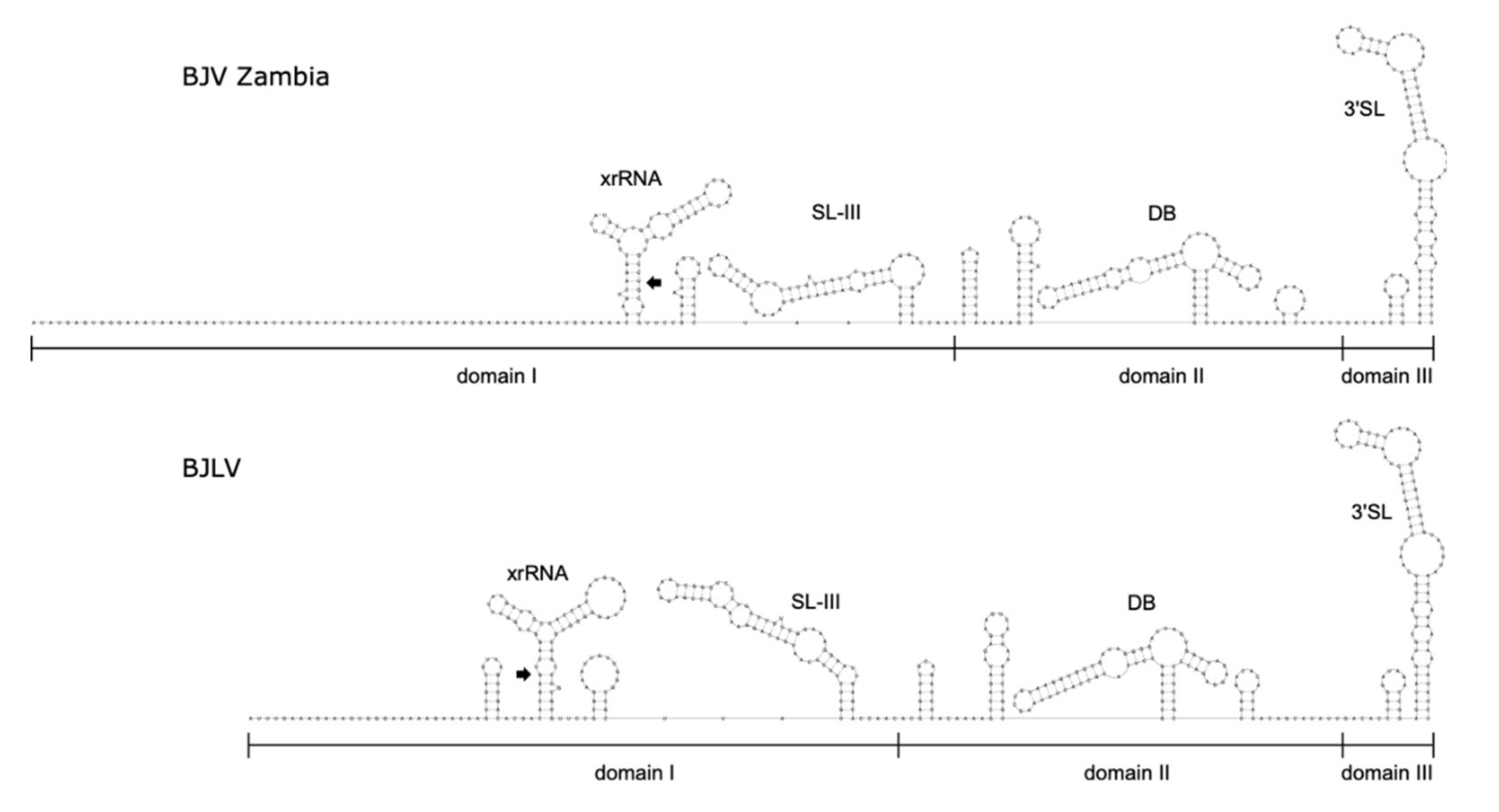
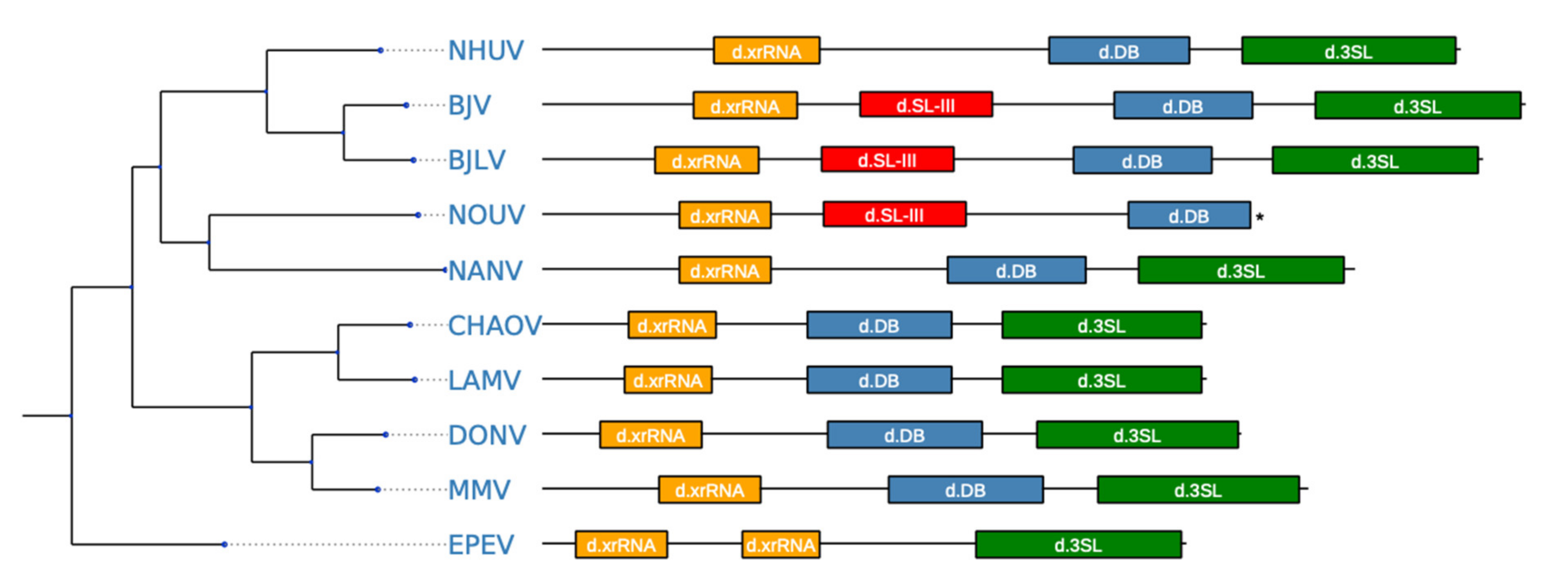
3.6. Secondary Structure Analysis of 3′-UTRs
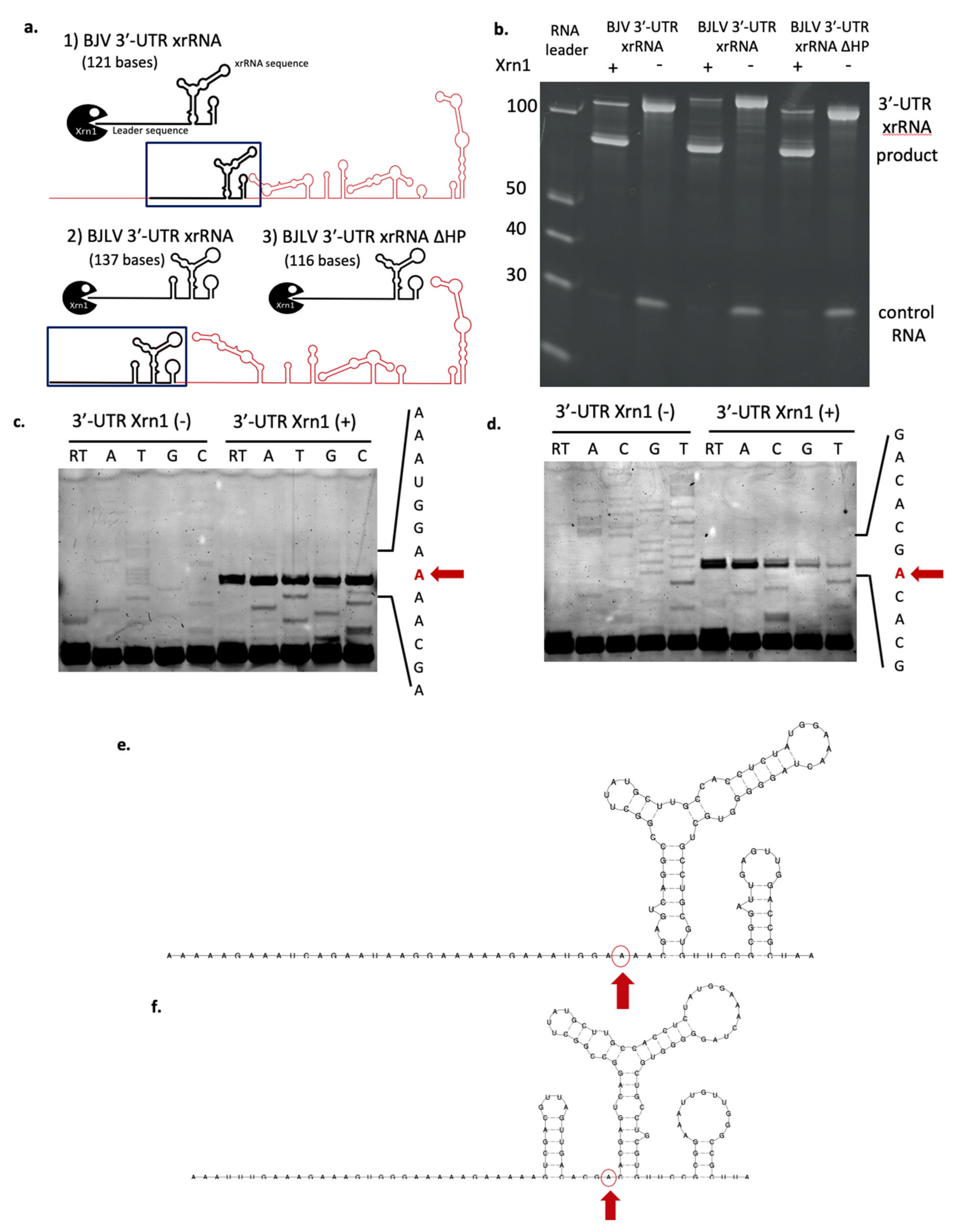
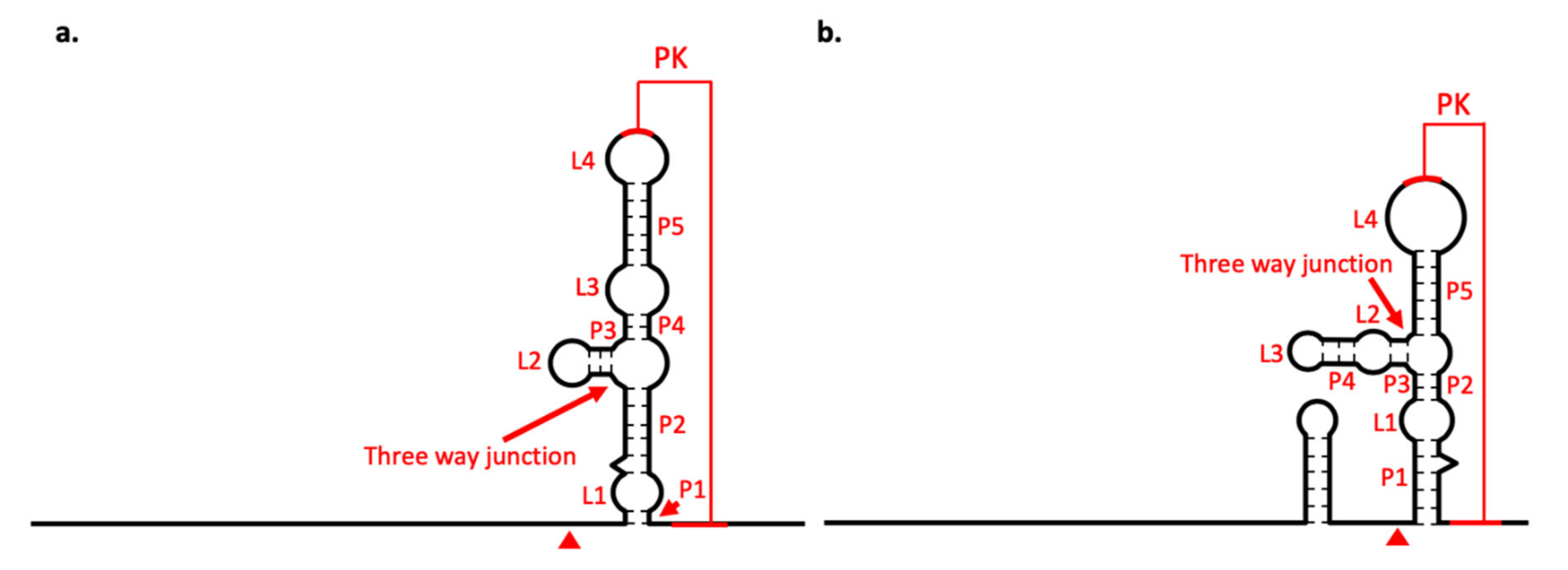
3.7. xrRNA Structure of BJV and BJLV Can Block Xrn1 Enzyme
3.8. Determination of Xrn1 Enzyme Stop Site
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bolling, B.G.; Weaver, S.C.; Tesh, R.B.; Vasilakis, N. Insect-Specific Virus Discovery: Significance for the Arbovirus Community. Viruses 2015, 7, 4911–4928. [Google Scholar] [CrossRef] [Green Version]
- Blitvich, B.J.; Firth, A.E. A Review of Flaviviruses that Have No Known Arthropod Vector. Viruses 2017, 9, 154. [Google Scholar] [CrossRef] [Green Version]
- Huhtamo, E.; Moureau, G.; Cook, S.; Julkunen, O.; Putkuri, N.; Kurkela, S.; Uzcátegui, N.Y.; Harbach, R.E.; Gould, E.A.; Vapalahti, O.; et al. Novel insect-specific flavivirus isolated from northern Europe. Virology 2012, 433, 471–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman, H.; Contreras-Gutierrez, M.A.; Travassos da Rosa, A.P.A.; Nunes, M.R.T.; Cardoso, J.F.; Popov, V.L.; Young, K.I.; Savit, C.; Wood, T.G.; Widen, S.G.; et al. Characterization of Three New Insect-Specific Flaviviruses: Their Relationship to the Mosquito-Borne Flavivirus Pathogens. Am. J. Trop. Med. Hyg. 2018, 98, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Kenney, J.L.; Solberg, O.D.; Langevin, S.A.; Brault, A.C. Characterization of a novel insect-specific flavivirus from Brazil: Potential for inhibition of infection of arthropod cells with medically important flaviviruses. J. Gen. Virol. 2014, 95, 2796–2808. [Google Scholar] [CrossRef] [PubMed]
- Romo, H.; Kenney, J.L.; Blitvich, B.J.; Brault, A.C. Restriction of Zika virus infection and transmission in Aedes aegypti mediated by an insect-specific flavivirus. Emerg. Microbes Infect. 2018, 7, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Gao, X.; Liang, G. Newly recognized mosquito-associated viruses in mainland China, in the last two decades. Virol. J. 2011, 8, 68. [Google Scholar] [CrossRef] [Green Version]
- Huhtamo, E.; Putkuri, N.; Kurkela, S.; Manni, T.; Vaheri, A.; Vapalahti, O.; Uzcátegui, N.Y. Characterization of a novel flavivirus from mosquitoes in northern europe that is related to mosquito-borne flaviviruses of the tropics. J. Virol. 2009, 83, 9532–9540. [Google Scholar] [CrossRef] [Green Version]
- Pauvolid-Corrêa, A.; Solberg, O.; Couto-Lima, D.; Kenney, J.; Serra-Freire, N.; Brault, A.; Nogueira, R.; Langevin, S.; Komar, N. Nhumirim virus, a novel flavivirus isolated from mosquitoes from the Pantanal, Brazil. Arch. Virol. 2015, 160, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Kolodziejek, J.; Pachler, K.; Bin, H.; Mendelson, E.; Shulman, L.; Orshan, L.; Nowotny, N. Barkedji virus, a novel mosquito-borne flavivirus identified in Culex perexiguus mosquitoes, Israel, 2011. J. Gen. Virol. 2013, 94, 2449–2457. [Google Scholar] [CrossRef]
- Ryan, S.J.; Carlson, C.J.; Mordecai, E.A.; Johnson, L.R. Global expansion and redistribution of Aedes-borne virus transmission risk with climate change. PLoS Negl. Trop. Dis. 2019, 13, e0007213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samy, A.M.; Elaagip, A.H.; Kenawy, M.A.; Ayres, C.F.; Peterson, A.T.; Soliman, D.E. Climate Change Influences on the Global Potential Distribution of the Mosquito Culex quinquefasciatus, Vector of West Nile Virus and Lymphatic Filariasis. PLoS ONE 2016, 11, e0163863. [Google Scholar] [CrossRef] [PubMed]
- Alaniz, A.J.; Carvajal, M.A.; Bacigalupo, A.; Cattan, P.E. Global spatial assessment of Aedes aegypti and Culex quinquefasciatus: A scenario of Zika virus exposure. Epidemiol. Infect. 2018, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef]
- Villordo, S.M.; Carballeda, J.M.; Filomatori, C.V.; Gamarnik, A.V. RNA Structure Duplications and Flavivirus Host Adaptation. Trends Microbiol. 2016, 24, 270–283. [Google Scholar] [CrossRef] [Green Version]
- Parsch, J.; Braverman, J.M.; Stephan, W. Comparative sequence analysis and patterns of covariation in RNA secondary structures. Genetics 2000, 154, 909–921. [Google Scholar]
- Bernhart, S.H.; Hofacker, I.L. From consensus structure prediction to RNA gene finding. Brief Funct. Genom. Proteom. 2009, 8, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Filomatori, C.V.; Lodeiro, M.F.; Alvarez, D.E.; Samsa, M.M.; Pietrasanta, L.; Gamarnik, A.V. A 5′ RNA element promotes dengue virus RNA synthesis on a circular genome. Genes Dev. 2006, 20, 2238–2249. [Google Scholar] [CrossRef] [Green Version]
- Lodeiro, M.F.; Filomatori, C.V.; Gamarnik, A.V. Structural and functional studies of the promoter element for dengue virus RNA replication. J. Virol. 2009, 83, 993–1008. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.G.; Lim, X.N.; Ng, W.C.; Sim, A.Y.L.; Poh, H.X.; Shen, Y.; Lim, S.Y.; Sundstrom, K.B.; Sun, X.; Aw, J.G.; et al. Structure mapping of dengue and Zika viruses reveals functional long-range interactions. Nat. Commun. 2019, 10, 1408. [Google Scholar] [CrossRef] [Green Version]
- Ochsenreiter, R.; Hofacker, I.L.; Wolfinger, M.T. Functional RNA Structures in the 3′UTR of Tick-Borne, Insect-Specific and No-Known-Vector Flaviviruses. Viruses 2019, 11, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slonchak, A.; Khromykh, A.A. Subgenomic flaviviral RNAs: What do we know after the first decade of research. Antivir. Res. 2018, 159, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ministry of Lands and Natural Resources, R.o.Z. National Policy on Wetlands. Available online: https://www.mlnr.gov.zm/?wpfb_dl=50 (accessed on 23 November 2019).
- Andy, H.; Georgina, E.; Donall, C.E.; Pete, B.; Francis, L.; Jakob, S.; Andrew, S.; Douglas, S.; Mark, S.; Tom, W.; et al. Automatic detection of open and vegetated water bodies using sentinel 1 to map African malaria vector mosquito breeding habitats. Remote Sens. 2019, 11, 593. [Google Scholar] [CrossRef] [Green Version]
- Babaniyi, O.; Mazaba-Liwewe, M.L.; Masaninga, F.; Mwaba, P.; Mulenga, D.; Songolo, P.; Mweene-Ndumba, I.; Rudatsikira, E.; Siziya, S. Prevalence of Yellow Fever in North-Western Province of Zambia. Int. J. Public Health Epidemiol. 2016, 8, 29–32. [Google Scholar]
- Babaniyi, O.A.; Mwaba, P.; Songolo, P.; Mazaba-Liwewe, M.L.; Mweene-Ndumba, I.; Masaninga, F.; Rudatsikira, E.; Siziya, S. Seroprevalence of Zika virus infection specific IgG in Western and North-Western Provinces of Zambia. Int. J. Public Health Epidemiol. 2015, 4, 110–114. [Google Scholar]
- Mweene-Ndumba, I.; Siziya, S.; Monze, M.; Mazaba, M.L.; Masaninga, F.; Songolo, P.; Mwaba, P.; Babaniyi, O.A. Seroprevalence of West Nile Virus specific IgG and IgM antibodies in North-Western and Western provinces of Zambia. Afr. Health Sci. 2015, 15, 803–809. [Google Scholar] [CrossRef]
- Mazaba-Liwewe, M.L.; Siziya, S.; Monze, M.; Mweene-Ndumba, I.; Masaninga, F.; Songolo, P.; Malama, C.; Chizema, E.; Mwaba, P.; Babaniyi, O.A. First sero-prevalence of dengue fever specific immunoglobulin G antibodies in Western and North-Western provinces of Zambia: A population based cross sectional study. Virol. J. 2014, 11, 135. [Google Scholar] [CrossRef]
- Wastika, C.E.; Sasaki, M.; Yoshii, K.; Anindita, P.D.; Hang’ombe, B.M.; Mweene, A.S.; Kobayashi, S.; Kariwa, H.; Carr, M.J.; Hall, W.W.; et al. Serological evidence of Zika virus infection in non-human primates in Zambia. Arch. Virol. 2019, 164, 2165–2170. [Google Scholar] [CrossRef]
- Orba, Y.; Hang’ombe, B.M.; Mweene, A.S.; Wada, Y.; Anindita, P.D.; Phongphaew, W.; Qiu, Y.; Kajihara, M.; Mori-Kajihara, A.; Eto, Y.; et al. First isolation of West Nile virus in Zambia from mosquitoes. Transbound Emerg. Dis. 2018, 65, 933–938. [Google Scholar] [CrossRef]
- Torii, S.; Orba, Y.; Hang’ombe, B.M.; Mweene, A.S.; Wada, Y.; Anindita, P.D.; Phongphaew, W.; Qiu, Y.; Kajihara, M.; Mori-Kajihara, A.; et al. Discovery of Mwinilunga alphavirus: A novel alphavirus in Culex mosquitoes in Zambia. Virus Res. 2018, 250, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Kent, R.J. The Mosquitoes of Macha, Zambia. Available online: http://www.jhsph.edu/malaria (accessed on 10 October 2012).
- Gillet, J. Common African Mosquitoes and their Medical Importance; William Heinemann Medical Books LTD: London, UK, 1972. [Google Scholar]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Patel, P.; Landt, O.; Kaiser, M.; Faye, O.; Koppe, T.; Lass, U.; Sall, A.A.; Niedrig, M. Development of one-step quantitative reverse transcription PCR for the rapid detection of flaviviruses. Virol. J. 2013, 10, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Pickett, B.E.; Greer, D.S.; Zhang, Y.; Stewart, L.; Zhou, L.; Sun, G.; Gu, Z.; Kumar, S.; Zaremba, S.; Larsen, C.N.; et al. Virus pathogen database and analysis resource (ViPR): A comprehensive bioinformatics database and analysis resource for the coronavirus research community. Viruses 2012, 4, 3209–3226. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R.; Durbin, R. RNA sequence analysis using covariance models. Nucleic Acids Res. 1994, 22, 2079–2088. [Google Scholar] [CrossRef] [Green Version]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [Green Version]
- Will, S.; Reiche, K.; Hofacker, I.L.; Stadler, P.F.; Backofen, R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 2007, 3, e65. [Google Scholar] [CrossRef]
- Bernhart, S.H.; Hofacker, I.L.; Will, S.; Gruber, A.R.; Stadler, P.F. RNAalifold: Improved consensus structure prediction for RNA alignments. BMC Bioinform. 2008, 9, 474. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Wolfinger, M.T.; Fallmann, J.; Eggenhofer, F.; Amman, F. ViennaNGS: A toolbox for building efficient next- generation sequencing analysis pipelines. F1000Res 2015, 4, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilweg, I.W.; Gultyaev, A.P.; Olsthoorn, R.C. Structural features of an Xrn1-resistant plant virus RNA. RNA Biol. 2019, 16, 838–845. [Google Scholar] [CrossRef] [Green Version]
- Chapman, E.G.; Moon, S.L.; Wilusz, J.; Kieft, J.S. RNA structures that resist degradation by Xrn1 produce a pathogenic Dengue virus RNA. Elife 2014, 3, e01892. [Google Scholar] [CrossRef] [PubMed]
- Kuno, G.; Chang, G.J.; Tsuchiya, K.R.; Karabatsos, N.; Cropp, C.B. Phylogeny of the genus Flavivirus. J. Virol. 1998, 72, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clyde, K.; Harris, E. RNA secondary structure in the coding region of dengue virus type 2 directs translation start codon selection and is required for viral replication. J. Virol. 2006, 80, 2170–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clyde, K.; Barrera, J.; Harris, E. The capsid-coding region hairpin element (cHP) is a critical determinant of dengue virus and West Nile virus RNA synthesis. Virology 2008, 379, 314–323. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.B.; Ochsenreiter, R.; Hostager, R.; Hofacker, I.L.; Janies, D.; Wolfinger, M.T. Updated Phylogeny of Chikungunya Virus Suggests Lineage-Specific RNA Architecture. Viruses 2019, 11, 798. [Google Scholar] [CrossRef] [Green Version]
- Funk, A.; Truong, K.; Nagasaki, T.; Torres, S.; Floden, N.; Balmori Melian, E.; Edmonds, J.; Dong, H.; Shi, P.Y.; Khromykh, A.A. RNA structures required for production of subgenomic flavivirus RNA. J. Virol. 2010, 84, 11407–11417. [Google Scholar] [CrossRef] [Green Version]
- Pijlman, G.P.; Funk, A.; Kondratieva, N.; Leung, J.; Torres, S.; van der Aa, L.; Liu, W.J.; Palmenberg, A.C.; Shi, P.Y.; Hall, R.A.; et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 2008, 4, 579–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roby, J.A.; Pijlman, G.P.; Wilusz, J.; Khromykh, A.A. Noncoding subgenomic flavivirus RNA: Multiple functions in West Nile virus pathogenesis and modulation of host responses. Viruses 2014, 6, 404–427. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, B.M.; Laurence, H.M.; Massey, A.R.; Costantino, D.A.; Xie, X.; Yang, Y.; Shi, P.Y.; Nix, J.C.; Beckham, J.D.; Kieft, J.S. Zika virus produces noncoding RNAs using a multi-pseudoknot structure that confounds a cellular exonuclease. Science 2016, 354, 1148–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sztuba-Solinska, J.; Teramoto, T.; Rausch, J.W.; Shapiro, B.A.; Padmanabhan, R.; Le Grice, S.F. Structural complexity of Dengue virus untranslated regions: Cis-acting RNA motifs and pseudoknot interactions modulating functionality of the viral genome. Nucleic Acids Res. 2013, 41, 5075–5089. [Google Scholar] [CrossRef] [PubMed]
- MacFadden, A.; O’Donoghue, Z.; Silva, P.A.G.C.; Chapman, E.G.; Olsthoorn, R.C.; Sterken, M.G.; Pijlman, G.P.; Bredenbeek, P.J.; Kieft, J.S. Mechanism and structural diversity of exoribonuclease-resistant RNA structures in flaviviral RNAs. Nat. Commun. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, A.B.; Wolfinger, M.T. Musashi binding elements in Zika and related Flavivirus 3′UTRs: A comparative study in silico. Sci. Rep. 2019, 9, 6911. [Google Scholar] [CrossRef] [Green Version]
- Brinton, M.A. Replication cycle and molecular biology of the West Nile virus. Viruses 2013, 6, 13–53. [Google Scholar] [CrossRef] [Green Version]
- Ndiaye, E.H.; Diallo, D.; Fall, G.; Ba, Y.; Faye, O.; Dia, I.; Diallo, M. Arboviruses isolated from the Barkedji mosquito-based surveillance system, 2012–2013. BMC Infect. Dis. 2018, 18, 642. [Google Scholar] [CrossRef]
- Camp, J.V.; Karuvantevida, N.; Chouhna, H.; Safi, E.; Shah, J.N.; Nowotny, N. Mosquito biodiversity and mosquito-borne viruses in the United Arab Emirates. Parasit Vectors 2019, 12, 153. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Rejon, J.E.; Blitvich, B.J.; Farfan-Ale, J.A.; Loroño-Pino, M.A.; Chi Chim, W.A.; Flores-Flores, L.F.; Rosado-Paredes, E.; Baak-Baak, C.; Perez-Mutul, J.; Suarez-Solis, V.; et al. Host-feeding preference of the mosquito, Culex quinquefasciatus, in Yucatan State, Mexico. J. Insect Sci. 2010, 10, 32. [Google Scholar] [CrossRef] [Green Version]
- Junglen, S.; Kopp, A.; Kurth, A.; Pauli, G.; Ellerbrok, H.; Leendertz, F.H. A new flavivirus and a new vector: Characterization of a novel flavivirus isolated from uranotaenia mosquitoes from a tropical rain forest. J. Virol. 2009, 83, 4462–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göertz, G.P.; van Bree, J.W.M.; Hiralal, A.; Fernhout, B.M.; Steffens, C.; Boeren, S.; Visser, T.M.; Vogels, C.B.F.; Abbo, S.R.; Fros, J.J.; et al. Subgenomic flavivirus RNA binds the mosquito DEAD/H-box helicase ME31B and determines Zika virus transmission by Aedes aegypti. Proc. Natl. Acad. Sci. USA 2019, 116, 19136–19144. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Month | Place | Species | Mosquito no. | Pool no. | No. of Flavivirus Positive Pools (%) * | Nucleotide Identity with BJV Israel |
|---|---|---|---|---|---|---|---|
| 2017 | April | Livingstone | Culex quinquefasciatus | 780 | 34 | 1 (2.9) a | 99% |
| Culex univittatus + | 15 | 3 | |||||
| Culex nebulosus + | 5 | 3 | |||||
| Culex tigripes + | 11 | 3 | |||||
| Culex bitaeniorhynchus + | 1 | 1 | |||||
| Aedes aegypti | 5 | 5 | |||||
| Aedes spp. + | 7 | 3 | |||||
| Mansonia sp. | 2 | 2 | |||||
| Anopheles spp. + | 72 | 15 | |||||
| Total | 898 | 69 | 1 | ||||
| 2017 | May | Mongu | Culex quinquefasciatus | 285 | 22 | ||
| Culex univittatus + | 309 | 15 | 2 (13.3) b | 81% | |||
| Culex annulioris + | 41 | 5 | 1 (20) c | 81% | |||
| Culex tigripes + | 2 | 2 | |||||
| Culex spp. + | 109 | 5 | |||||
| Aedes macintoshi + | 10 | 3 | |||||
| Aedes aegypti | 1 | 1 | |||||
| Coquillettidia aurites + | 9 | 2 | |||||
| Coquillettidia metarika + | 7 | 2 | |||||
| Coquillettidia fuscopenata + | 46 | 5 | |||||
| Mansonia sp. | 132 | 8 | |||||
| Uranotaenia sp. + | 1 | 1 | |||||
| Aedeomya sp. | 10 | 2 | |||||
| Anopheles spp. + | 1444 | 65 | |||||
| Total | 2406 | 138 | 3 |
| Virus | C/Anch C | Anch C/prM | PrM/M | M/E |
| BJV Israel | KTSKR/GLQQS | TMAAC/ATLGM | RRSKR/SVAIA | APAYS/LHCSR |
| BJV Oman | KTSKR/GLQQS | TMAAC/ATLGM | RRSKR/SVAIA | APAYS/LHCSR |
| BJV Zambia | KTSKR/GLQQS | TMAAC/ATLGM | RRSKR/SVAIA | APAYS/LHCSR |
| BJLV | RTAKR/GLGQS | TMAAC/ATLGM | RRSKR/SVAIA | APAYS/LHCAR |
| Virus | E/NS1 | NS1/NS2A | NS2A/NS2B | NS2B/NS3 |
| BJV Israel | TTVAG/DVGCN | SWTTA/GNATG | GSGKR/SVSMG | KGTQK/AGAMW |
| BJV Oman | TTVAG/DVGCN | SWTTA/GNATG | GSGKR/SVSMG | KGTQK/AGAMW |
| BJV Zambia | TTVAG/DVGCN | SWTTA/GNATG | GSGKR/SVSMG | KGTQK/AGAMW |
| BJLV | TTVAG/DVGCN | SWSTA/GNISG | AAGRR/SVSMG | KPAQK/AGAMW |
| Virus | NS3/NS4A | NS4A/2K | 2K/NS4B | NS4B/NS5 |
| BJV Israel | AEGRR/GASDI | AEKQR/SAIDN | LAVTA/NEKGL | KSARK/GTPGG |
| BJV Oman | AEGRR/GASDI | AEKQR/SAIDN | LAVTA/NEKGL | KSARK/GTPGG |
| BJV Zambia | AEGRR/GASDI | AEKQR/SAIDN | LAVTA/NEKGL | KSARK/GTPGG |
| BJLV | AEGRR/GAHDL | AEKQR/SAIDN | LAVTA/NEKGL | KSARK/GTPGG |
| Virus | AnchC-C | prM | M | Envelope | NS1 | NS2A | ||||||
| aa | ID (%) | aa | ID (%) | aa | ID (%) | aa | ID (%) | aa | ID (%) | aa | ID (%) | |
| BJLV | 127 | - | 92 | - | 75 | - | 503 | - | 350 | - | 233 | - |
| BJV Zambia | 125 | 86.6 | 992 | 91.3 | 75 | 88.0 | 503 | 90.4 | 350 | 92.2 | 233 | 81.5 |
| Nhumirim virus | 128 | 67.9 | 94 | 64.5 | 75 | 72.0 | 503 | 64.7 | 351 | 71.4 | 232 | 66.8 |
| Virus | NS2B | NS3 | NS4A | NS4B | NS5 | TOTAL | ||||||
| aa | ID (%) | aa | ID (%) | aa | ID (%) | aa | ID (%) | aa | ID (%) | aa | ID (%) | |
| BJLV | 129 | - | 621 | - | 126 | - | 255 | - | 905 | - | 3439 | - |
| BJV Zambia | 129 | 90.6 | 621 | 92.4 | 126 | 84.9 | 255 | 91.7 | 905 | 92.1 | 3437 | 90.6 |
| Nhumirim virus | 130 | 63.9 | 622 | 74.4 | 149 | 62.6 | 255 | 73.7 | 907 | 77.4 | 3446 | 71.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wastika, C.E.; Harima, H.; Sasaki, M.; Hang’ombe, B.M.; Eshita, Y.; Qiu, Y.; Hall, W.W.; Wolfinger, M.T.; Sawa, H.; Orba, Y. Discoveries of Exoribonuclease-Resistant Structures of Insect-Specific Flaviviruses Isolated in Zambia. Viruses 2020, 12, 1017. https://doi.org/10.3390/v12091017
Wastika CE, Harima H, Sasaki M, Hang’ombe BM, Eshita Y, Qiu Y, Hall WW, Wolfinger MT, Sawa H, Orba Y. Discoveries of Exoribonuclease-Resistant Structures of Insect-Specific Flaviviruses Isolated in Zambia. Viruses. 2020; 12(9):1017. https://doi.org/10.3390/v12091017
Chicago/Turabian StyleWastika, Christida E., Hayato Harima, Michihito Sasaki, Bernard M. Hang’ombe, Yuki Eshita, Yongjin Qiu, William W. Hall, Michael T. Wolfinger, Hirofumi Sawa, and Yasuko Orba. 2020. "Discoveries of Exoribonuclease-Resistant Structures of Insect-Specific Flaviviruses Isolated in Zambia" Viruses 12, no. 9: 1017. https://doi.org/10.3390/v12091017
APA StyleWastika, C. E., Harima, H., Sasaki, M., Hang’ombe, B. M., Eshita, Y., Qiu, Y., Hall, W. W., Wolfinger, M. T., Sawa, H., & Orba, Y. (2020). Discoveries of Exoribonuclease-Resistant Structures of Insect-Specific Flaviviruses Isolated in Zambia. Viruses, 12(9), 1017. https://doi.org/10.3390/v12091017